Introduction
Heart failure is the leading cause of heart
disease-associated death worldwide and is caused by various
pathologies, including myocardial infarction and hypertension
(1). Heart failure is one of the
most common causes of atrial fibrillation (AF), which induces
atrial electrical and structural remodeling, as well as increased
atrial fibrosis (2). Atrial fibrosis
contributes to the development and persistence of AF and thus plays
a role in the AF-induced impairment of electrical conduction
between cardiomyocytes, leading to reduced conduction velocity and
increased conduction dispersion (3).
Glucagon-like peptide-1 (GLP-1), an incretin
released postprandially, has been shown to exert cardioprotective
effects in addition to glycemic regulatory effects (4). Following its release, GLP-1 is quickly
degraded by dipeptidyl peptidase 4 (DPP-4). GLP-1 receptor
activation preserves cardiac performance by activating multiple
pro-survival kinases and modulating cell apoptosis and energy
utilization (5-7),
and exendin-4 (a long-acting GLP-1 analogue)-mediated GLP-1
receptor activation in models of MI inhibits cardiac remodeling by
mitigating oxidative stress-induced injury (6) and attenuating interstitial fibrosis
(4). GLP-1 receptors are expressed
in atrial myocytes and arteries (8).
Previous studies have focused mainly on ventricular structural
remodeling (6,9). Thus, whether exendin-4 influences
atrial arrhythmogenesis in a rat heart failure model remains
unknown.
The acute activation of the AKT signaling pathway, a
survival signaling pathway, protects the heart from
ischemia/reperfusion injury (10).
In contrast, the long-term activation of the AKT signaling pathway
leads to cardiac hypertrophy and fibrosis (11). A previous study demonstrated that
GLP-1 receptor activation protected rat hearts from
ischemia/reperfusion injury via the activation of the AKT pathway
(12). To better understand the
association between the GLP-1 receptor and atrial fibrosis in heart
failure, the present study aimed to evaluate the effects of
exendin-4 treatment on atrial fibrosis and arrhythmia vulnerability
in a rat model of MI-induced heart failure and to investigate the
mechanisms underlying these effects.
Materials and methods
Experimental animals
The experiments were performed in strict accordance
with the recommendation in the Guide for the Care and Use of
Laboratory Animals from the Institute for Laboratory Animal
Research, National Research Council, Washington, D.C., National
Academy Press, 2011(13). Moreover,
all experimental protocols were approved by the Experimental Animal
Care and Institutional Animal Ethical Committee of Guizhou Medical
University (Guiyang, China).
In total, 88 male Sprague-Dawley rats weighting
between 200-250 g, were purchased from Hunan Silaike Jingda
Laboratory Animal Co., Ltd. The rats were acclimatized at 25˚C with
a 12-h light/dark cycle and relative humidity of between 40 and
70%, and had free access to food and water.
The rats were randomly divided into the following
four groups: i) A sham-operated group comprising of healthy rats
without heart failure that received 4 weeks of intraperitoneal
saline treatment; ii) a myocardial infarction group (MI group)
comprising of rats with coronary ligation-induced heart failure
that received 4 weeks of intraperitoneal saline treatment; iii) an
MI plus Exendin-4 group (MI+Ex-4 group) comprising of rats with
heart failure that received an intraperitoneal injection of 10
µg/kg/day (14) exendin-4 (the dose
was selected based on the results of a previous study by the
authors) (9) for 4 weeks and iv) an
MI plus exendin9-39 and exendin-4 group (MI+Ex-4+Ex9-39 group)
comprising of rats with heart failure that received an
intraperitoneal injection of 150 µg/kg/day (15) exendin9-39 and 10 µg/kg/day exendin-4
for 4 weeks. Each group comprised 22 rats. The drugs and saline
were administered beginning at 24 h after surgery, and 1/2 of the
total dose was administered every 12 h. The experimental groups and
treatment schedules are presented in Fig. 1.
Coronary ligation
The coronary ligation procedure performed herein was
described in a previous study by the authors (16). The rats were anesthetized with an
intraperitoneal injection of pentobarbital sodium (40 mg/kg,
Sigma-Aldrich; Merck KGaA). After full anesthesia was achieved when
pain reflex disappeared, artificial respiration was initiated using
a volume-constant rodent ventilator. The chest was opened through a
left thoracotomy at the third intercostal space, after which left
anterior descending (LAD) artery ligation was performed using a 7-0
silk suture. The rats in the sham-operated group underwent the same
surgical procedure, but did not undergo coronary artery
ligation.
Echocardiographic measurements
Echocardiography (Vevo 770; Visual Sonics) was
performed under anesthesia with an intraperitoneal injection of
pentobarbital sodium (40 mg/kg, Sigma-Aldrich; Merck KGaA) to
assess the left ventricular function 4 weeks after the
administration of saline/Ex-4/Ex-4+Ex9-39. Atrial analyses included
left atrial (LA) dimension at end diastole and systole and LA
fractional shortening. A pulse-wave Doppler was used to assess
mitral valve flow as an index of diastolic function.
Hemodynamic measurements
Systemic hemodynamic analyses were measured with 2.0
F Millar catheters inserted into the right carotid artery and then
advanced into the left ventricle in rats anesthetized with an
intraperitoneal injection of pentobarbital sodium (40 mg/kg,
Sigma-Aldrich; Merck KGaA). Following the hemodynamic measurements,
the rats were euthanized by cervical dislocation under the
aforementioned anesthesia. The lungs and tibiae of the rats were
dissected and measured for the lung weight/tibia length (LW/TL,
mg/mm) ratio calculation.
Preparation of Langendorff-perfused
hearts
The rats (n=12 in each group) were anesthetized as
aforementioned, heparinized (heparin sodium, 400 U, i.p.,
Sigma-Aldrich; Merck KGaA). Their hearts were rapidly harvested,
rinsed and perfused in a Langendorff perfusion system
(ADInstruments) with oxygenated HEPES-buffered Tyrode solution
containing the following components (mmol/l): NaCl, 135; KCl, 5.4;
CaCl2, 1.8; MgCl2, 1; Na2HPO4,
0.3; HEPES, 10 and glucose, 10 (pH 7.3) at a constant pressure
(70-90 cm H2O) and temperature (37.0±0.5˚C). The hearts
were perfused for 20 min prior to microelectrode array recording,
monophasic action potential (MAP) recordings and atrial
tachyarrhythmia susceptibility.
Microelectrode array (MEA)
recording
MEA recording was conducted on the
Langendorff-perfused hearts for extracellular electrophysiological
analysis and excitation propagation observations, which were
performed 4 weeks after MI. An array of 32 monopolar electrodes (32
Map) capable of covering a 3x3-mm square (inter-electrode distance
of 500 µm) was placed on the left atrial appendage (LAA). The 32
Map signals were obtained and processed using Cardio 2D_2.6.2 and
Cardio 2D+_2.4.2 software (Multi Channel Systems MCS GmbH). The
total activation time (TAT) was defined as the longest possible
period in which the excitation was passed from the first excited
electrode to the last excited electrode. The TAT difference of each
heartbeat was measured for the TAT dispersion as an index of
conduction homogeneity. TAT dispersion was estimated by the
standard deviation of the total activation time in the mapped area
in 60 sec. Conduction velocity was assessed by linear regression
analysis of the isochrones distance vs. activation time in MEA
recording (17).
MAP recordings and atrial
tachyarrhythmia susceptibility
A pair of platinum stimulating electrodes were
placed on the epicardial surface of the right atrial appendage for
programmed stimulation, and custom-made-Ag-AgCl electrodes
consisting of two 0.25-mm Teflon-coated silver wires were
positioned on the LAA for MAP recording. The 20, 50 and 90 action
potential durations (APD20, APD50 and
APD90) were calculated at a pacing cycle length (PCL) of
250 msec by performing at least eight successive MAPs. Burst pacing
(2-msec pulse width at 20 Hz, 2-sec burst duration) was used to
induce atrial tachyarrhythmias, which were defined as continuous
atrial contractions lasting >2 sec. Sustained atrial
tachyarrhythmia was defined as contractions lasting >30 sec.
Histological analysis
The rats were euthanized by cervical dislocation
under anesthesia (intraperitoneal pentobarbital sodium, 40 mg/kg,
Sigma-Aldrich; Merck KGaA) following the administration of the
drugs for 4 weeks. The hearts were excised and arrested in diastole
with 10% KCl, fixed in 10% buffered formalin at room temperature
for 24 h and embedded in paraffin. The tissues were subsequently
cut into 3-µm thick sections and stained with Masson's trichrome at
room temperature for 2 h to assess interstitial collagen
deposition. All heart sections were examined, and images were
captured via light microscopy (magnification, x100 and x400; BX-50;
Olympus Corporation). Data analysis was performed using Image-Pro
Plus 6.0 software (Media Cybernetics, Inc.). The collagen volume
fraction (CVF) was calculated as a percentage of the interstitial
collagen area.
Measurement of plasma GLP-1 and atrial
natriuretic peptide (ANP) concentrations
Plasma GLP-1 (n=6; cat. no. EZGLP1T-36K;
Sigma-Aldrich; Merck KGaA) and ANP (n=6; cat. no. E-EL-R0017;
Elabscience, Inc.) concentrations were assessed using post-caval
blood samples using enzyme immunoassay kits.
Western blotting
Total protein was extracted from the rat LAA tissue
using RIPA lysis buffer (Beyotime Institute of Biotechnology).
Protein concentration was examined using a Bicinchoninic Acid
Protein Assay kit (cat. no. AS1086; Wuhan Aspen Biotechnology Co.,
Ltd.). Protein samples (40 µg/lane) were loaded onto an 8-12% gel,
separated using SDS-PAGE and transferred onto polyvinylidene
difluoride membranes. The membranes were subsequently incubated
with primary antibodies against the GLP-1 receptor (cat. no.
ab111125; 1:1,000, Abcam), collagen I (Col I; cat. no. ab34710;
1:200; Abcam), collagen III (Col III; cat. no. ab184993; 1:2,000;
Abcam), transforming growth factor (TGF)-β1 (cat. no. ab179695;
1:600; Abcam), total AKT (1:1,000, Abcam, ab8805), phosphorylated
AKT (cat. no. ab38449; 1:2,000, Abcam), total PI3K (cat. no.
ab154598; 1:1,000, Abcam) and phosphorylated PI3K (cat. no.
ab191606; 1:200; Abcam) overnight at 4˚C. Following which,
membranes were incubated with horseradish peroxidase
(HRP)-conjugated rabbit anti-goat immunoglobulin G (IgG) (cat. no.
AS1108; 1:10,000; Wuhan Aspen Biotechnology Co., Ltd.) or
HRP-conjugated goat anti-rabbit IgG (cat. no. AS1107; 1:10,000;
Wuhan Aspen Biotechnology Co., Ltd.) secondary antibodies for 2 h
at room temperature. Chemiluminescence reagent (ECL kit; Beyotime
Institute of Biotechnology) was used to visualize the proteins, and
quantitative densitometric analysis was performed using Image-Pro
Plus 6.0 software (Media Cybernetics, Inc.).
Reverse transcription-quantitative PCR
(RT-qPCR)
RT-qPCR was used to analyze GLP-1 receptor
mRNA expression. Total RNA was isolated from the LAA tissues using
a Total RNA Extraction kit (RN0401; Aidlab Biotechnologies Co.,
Ltd.). RNA samples (4 µg) were reverse transcribed into cDNA using
the Transcriptor First Strand cDNA Synthesis kit (cat. no. R101-01;
Vazyme Biotech Co., Ltd.). The reverse transcription temperature
protocol was as follows: 25˚C for 5 min, 50˚C for 15 min, 85˚C for
5 min and 4˚C for 10 min. qPCR was performed using using SYBR Green
master mix (Roche Diagnostics). The following thermocycling
conditions were used for the qPCR: 50˚C for 2 min, 95˚C for 10 min;
followed by 40 cycles of denaturation at 95˚C for 30 sec, annealing
and extension at 60˚C for 30 sec. The data were analyzed using the
2-ΔΔCq method (18). GLP-1 receptor mRNA expression levels
were normalized to GAPDH expression levels. The following primers
used were used in the present study: GAPDH, forward:
5'-ACAGCAACAGGGTGGTGGAC-3' and reverse: 5'-TTTGAGGGTGCAGCGAACTT-3;
and GLP-1 receptor, forward: 5'-TCTTCTGCAACCGAACCTTT-3' and
reverse: 5'-TGCCCTTGGAGCACACTACT-3.
Statistical analysis
Statistical analysis was performed using SPSS 22.0
software (IBM Corp). All experiments were independently performed
three times. Numerical data are expressed as the means ± standard
error of the mean and were analyzed using one-way analysis of
variance (ANOVA), followed by the Bonferroni's multiple comparison
test. Categorical data, which included ATA incidence and sustained
ATA incidence, were analyzed by the χ2 test and Fisher's
exact test. P<0.05 was considered to indicate a statistically
significant difference.
Results
Exendin-4 improves heart function
following MI
Post-MI left atrium dimensions were increased and
atrial fractional shortening decreased compared with sham rats
(Fig. 2A). Furthermore, the LW/TL
ratios and plasma ANP concentration were increased following MI
(Fig. 2B). Hemodynamic measurements
revealed reduced contractility (dP/dtmax) and relaxation (dP/dtmin)
in the rats with MI compared with sham rats (Fig. 2C). These abnormalities were
attenuated by exendin-4 treatment. Moreover, the co-administration
of exendin9-39 and exendin-4 diminished the protective effects of
exendin-4 on cardiac function.
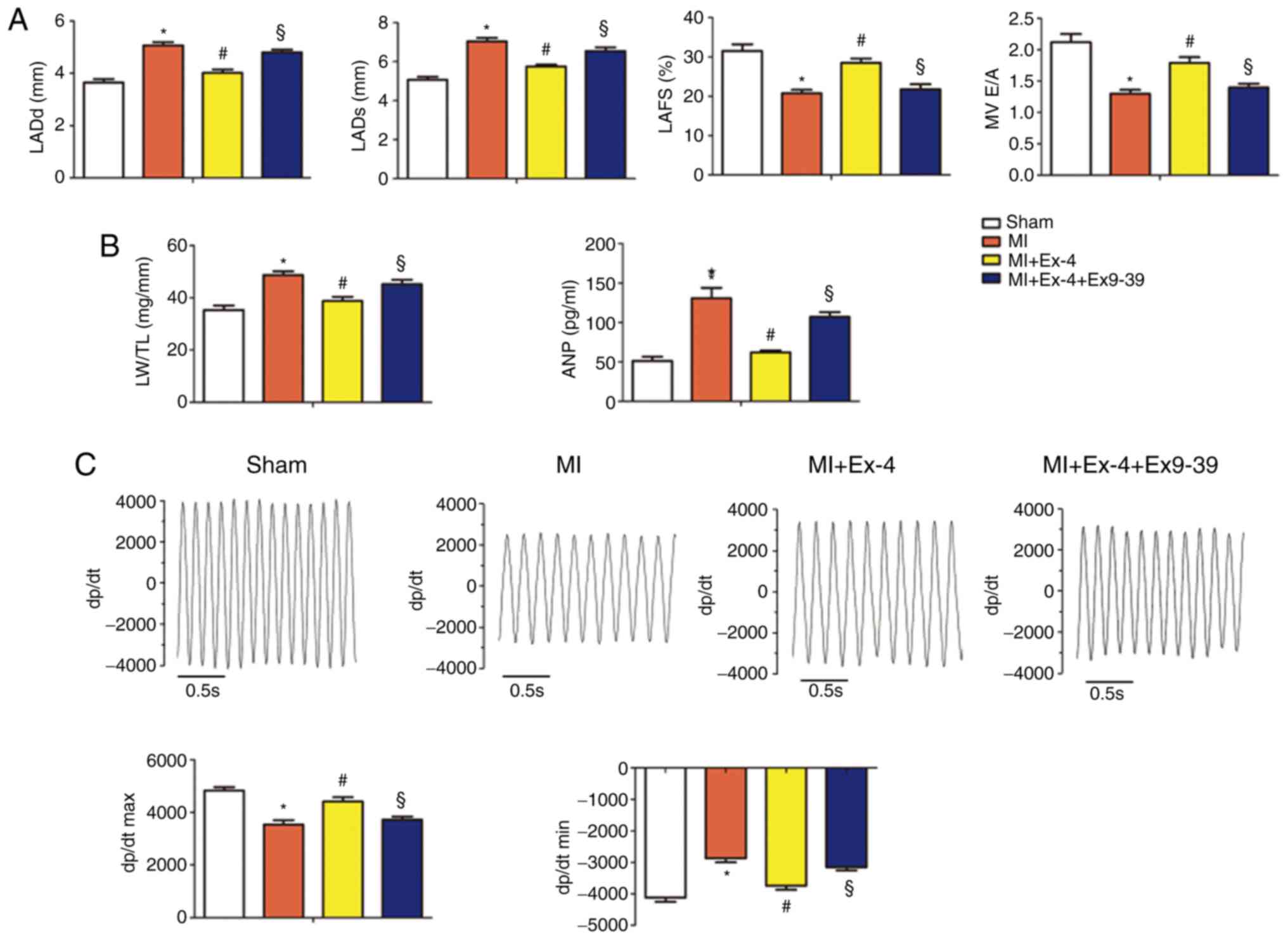 | Figure 2The GLP-1 receptor agonist,
exendin-4, preserves heart function and hemodynamics. (A)
Echocardiographic data for the sham-operated, MI, MI+Ex-4 and
MI+Ex-4+Ex9-39 groups (n=9 animals per group). MV E/A, LADd, LADs
and LAFS were analyzed using echocardiography. (B) LW/TL ratios and
the plasm levels of ANP in the sham-operated, MI, MI+Ex-4 and
MI+Ex-4+Ex9-39 groups (n=9 animals per group). (C) Representative
recordings of dp/dt and the statistical analyses of dP/dtmax,
dP/dtmin, SBP, DBP, MBP in the sham-operated, MI, MI+Ex-4 and
MI+Ex-4+Ex9-39 rat hearts at 4 weeks following sham or MI surgery
(n=9 animals per group). *P<0.05 vs. the
sham-operated group, #P<0.05 vs. the MI group,
§P<0.05 vs. the MI+Ex-4 group. MI, myocardial
infarction; Ex-4, exendin-4; Ex9-39, exendin9-39; LW/TL, lung
weight to tibia length ratio; MV E/A, mitral valve E/A; LADd, LA
dimension in ventricular diastole; LADs, LA dimension in
ventricular systole; LAFS, LA fractional shortening; ANP, atrial
natriuretic peptide; SBP, systolic blood pressure; DBP, diastolic
blood pressure; MBP, mean blood pressure. |
Exendin-4 ameliorates atrial
electrical remodeling in heart failure
To determine whether exendin-4 treatment exerts
effects on atrial electrophysiology, the APDs and atrial
tachyarrhythmia inducibility were measured in Langendorff-perfused
hearts. It was found that the APD50 and APD90
were prolonged following MI compared with sham rats; however, the
APD50 and APD90 were shorter in the exendin-4
group compared with the MI group (Fig.
3A). Additionally, atrial tachyarrhythmias occurred in two out
of 12 sham-operated hearts and 11/12 hearts with MI. However,
atrial tachyarrhythmias occurred in five out of 12 hearts treated
with exendin-4 (Fig. 3B).
Furthermore, the incidence of sustained atrial tachyarrhythmias
inducibility was decreased in the hearts treated with exendin-4
compared with the hearts with MI (Fig.
3B). The effects of exendin-4 on atrial electrophysiology were
abolished by the co-administration of exendin9-39.
 | Figure 3The GLP-1 receptor agonist,
exendin-4, improves atrial electrophysiological function
properties. (A) Representative recordings of action potentials and
quantitative analysis of the APD20, APD50 and
APD90 of LAA in the sham-operated, MI, MI+Ex-4 and
MI+Ex-4+Ex9-39 rat hearts at four weeks following sham or MI
surgery (n=12 animals per group). (B) Representative recordings of
burst pacing-induced atrial tachyarrhythmias in the sham-operated,
MI, MI+Ex-4 and MI+Ex-4+Ex9-39 rat hearts at four weeks following
sham or MI surgery and statistical analyses of these data (n=12
animals per group). *P<0.05 vs. the sham-operated
group, #P<0.05 vs. the MI group,
§P<0.05 vs. the MI+Ex-4 group. MI, myocardial
infarction; Ex-4, exendin-4; Ex9-39, exendin9-39; APD, action
potential duration; LAA, left atrial appendage. |
Exendin-4 modulates disordered
excitation transmission
Representative excitation transmission maps of the
surface of the LAA in each group are presented in Fig. 4A. In the rats in the sham-operated
group, propagation was conducted rapidly from the first excited
electrode to the last excited electrode. Following MI, the signal
proceeded slowly, a change accompanied by an increased total
activation time and a decreased conduction velocity compared with
sham rats (Fig. 4C). Exendin-4
treatment increased the conduction velocity and decreased the total
activation time compared with MI rats, which were indicative of
improvement in conduction (Fig. 4C).
Representative tracings for TAT dispersion in the four groups are
presented in Fig. 4B. Compared with
the sham-operated hearts, the hearts with MI exhibited prolonged
TAT dispersion (Fig. 4B and C). Exendin-4 treatment restored TAT
dispersion to a level similar to that of the control value
(Fig. 4B and C), and there was no significant difference
between sham rat and MI+Ex-4 rats in TAT dispersion. As shown in
Fig. 4A, the activation pattern was
uniform in the sham-operated hearts; however, the activation
pattern was more heterogeneous in the hearts with MI. Exendin-4
treatment ameliorated the anisotropic conduction properties.
However, the GLP-1R antagonist partly diminished the beneficial
effects of exendin-4 on excitation transmission.
 | Figure 4Ex-4 attenuates conduction slowing
and conduction heterogeneity in the left atrial appendage following
myocardial infarction. (A) Representative recordings of maps in
each group. (B) Representative long-term traces of
instantaneous-beat TATs. (C) Statistical analysis of conduction
velocity, total activation time and TAT dispersion in the
sham-operated, MI, MI+Ex-4 and MI+Ex-4+Ex9-39 rat hearts 4 weeks
following sham or MI surgery (n=5 animals per group).
*P<0.05 vs. the sham-operated group,
#P<0.05 vs. the MI group, §P<0.05
versus the MI+Ex-4 group. MI, myocardial infarction; Ex-4,
exendin-4; Ex9-39, exendin9-39; TAT, total activation time; CV,
conduction velocity. |
Exendin-4 suppresses atrial
fibrosis
As atrial fibrosis is one of the main atrial
structural alterations responsible for the maintenance and
progression of AF (19), the
expression levels of profibrotic markers in the atrium were
analyzed. Specifically, Col I, Col III and TGF-β1 expression levels
were examined using western blotting and the CVF was assessed by
Masson's trichrome staining. It was demonstrated that the Col I,
Col III and TGF-β1 protein expression levels, and the CVF, were
higher in the rats with MI compared with those who underwent the
sham operation (Fig. 5A and B). However, these increases were attenuated
by exendin-4 treatment (Fig. 5A and
B). The expression of Col I, Col III
and TGF-β1 protein, and the CVF were increased in MI+Ex-4+Ex9-39
rats compared with MI+Ex-4 rats. Thus, the inhibition of GLP-1R by
exendin9-39 partly diminished the suppressive effects of exendin-4
on atrial fibrosis (Fig. 5A and
B).
 | Figure 5Ex-4 attenuates atrial fibrosis and
fibrotic markers expression in the left atrial appendage. (A)
Interstitial fibrosis was evaluated using Masson's staining for
collagen and quantitative analysis of the CVF. (B) Representative
western blots of atrial tissue lysates probed for Col I, Col III
and TGF-β1 expression in the sham-operated, MI, MI+Ex-4 and
MI+Ex-4+Ex9-39 rat hearts 4 weeks following sham or MI surgery and
statistical analysis of the protein expression levels of these
proteins, which were normalized to those of GAPDH (n=5 animals per
group). *P<0.05 vs. the sham-operated group,
#P<0.05 vs. the MI group, §P<0.05 vs.
the MI+Ex-4 group. MI, myocardial infarction; Ex-4, exendin-4;
Ex9-39, exendin9-39; CVF, collagen volume fraction; Col, collagen;
TGF, transforming growth factor. |
Exendin-4 inhibits the PI3K/AKT
signaling pathway activity
To determine whether the GLP-1 receptor plays a role
in atrial arrhythmogenesis in the MI-induced heart failure model,
atrial GLP-1 receptor expression and plasma GLP-1 levels were
assessed using western blotting analysis and ELISA. The plasma
GLP-1 levels and atrial GLP-1 receptor mRNA and protein
expression levels were significantly downregulated following MI
(Fig. 6A and B). Exendin-4 treatment increased the plasma
GLP-1 levels, and the atrial GLP-1 receptor mRNA and protein
expression levels (Fig. 6A-C).
However, these effects were partly neutralized by co-treatment with
exendin9-39 (Fig. 6A-C).
 | Figure 6Exendin-4 inhibits the PI3K/AKT
signaling pathway. (A) Plasma GLP-1 levels and (B) GLP-1
receptor mRNA expression as determined by reverse
transcription-quantitative PCR in the sham-operated, MI, MI+Ex-4
and MI+Ex-4+Ex9-39 rat atrial tissues 4 weeks following sham or MI
surgery. (C) Representative western blots of atrial tissues probed
for GLP-1 receptor, total PI3K, phosphorylated PI3K, total AKT and
phosphorylated AKT in the sham-operated, MI, MI+Exendin-4 and
MI+Exendin-4+Ex9-39 rat hearts four weeks following sham or MI
surgery and statistical analysis of the expression levels of these
proteins, which were normalized to those of GAPDH (n=5 animals per
group). *P<0.05 vs. the sham-operated group,
#P<0.05 vs. the MI group, §P<0.05 vs.
the MI+Ex-4 group. MI, myocardial infarction; Ex-4, exendin-4;
Ex9-39, exendin9-39; GLP-1, glucagon-like peptide-1 receptor; p-,
phosphorylated; t-, total. |
To investigate the mechanisms underlying the effects
of exendin-4 on atrial fibrosis, the PI3K/AKT signaling pathway was
assessed. Compared with the atrial tissues from sham-operated rats,
the tissues from rats with MI exhibited increased phosphorylated
PI3K and AKT levels compared with sham rats, although the total
PI3K and AKT levels remained unaltered between the two groups. The
increased expression levels of phosphorylated PI3K and AKT were
attenuated by exendin-4 treatment (Fig.
6B and C), and all the effects
of exendin-4 were neutralized by the co-administration of
exendin9-39 (Fig. 6B and C).
Discussion
To the best of our knowledge, the present study is
the first to demonstrate the effects of exendin-4 on atrial
fibrosis and atrial arrhythmogenesis susceptibility. The results
demonstrated that in rats with MI-induced heart failure, atrial
fibrosis and atrial arrhythmogenesis susceptibility were increased.
In addition, increased atrial tachyarrhythmias susceptibility,
reductions in conduction velocity and increases in conduction
dispersion were observed. Treatment with exendin-4 attenuated
atrial fibrosis and atrial arrhythmogenesis susceptibility. The
co-administration of exendin9-39 and exendin-4 partly diminished
the protective effects of exendin-4. The increased atrial
arrhythmogenesis susceptibility was associated with atrial
fibrosis, which partially induced by the enhanced phosphorylation
of the PI3K/AKT signaling pathway, an effect that was abolished by
exendin-4. The beneficial effects of exendin-4 were attenuated by
the co-administration of the GLP-1 receptor antagonist,
exendin9-39, and exendin-4. Taken together, these findings
indicated that exendin-4 attenuates atrial fibrosis at least partly
through PI3K/AKT signaling pathway inactivation via the GLP-1
receptor and inhibits atrial arrhythmogenesis.
The cardioprotective effects of exendin-4 are well
known and a previous study by the authors demonstrated the
cardioprotective effects of exendin-4 on a rat model of chronic MI
through the endothelial nitric oxide synthase/cyclic guanosine
monophosphate/protein kinase G pathway (9). Our previous studies showed that
exendin-4 has direct electrophysiological effect on
electrocardiogram parameters including QRS, QT and QTc intervals
and calcium handling in ventricular arrhythmias (9,20).
However, limited information is available regarding the effects of
exendin-4 on atrial electrophysiology in heart failure. Previous
studies have primarily focused on the effects of exendin-4 on
ventricular structural remodeling (6,9). In the
present study, the effects of exendin-4 on atrial arrhythmogenesis
were investigated in a model of MI-induced heart failure.
Sitagliptin (a DPP-4 inhibitor) has been shown to attenuate
ventricular arrhythmias in rats with MI by inhibiting sympathetic
innervation (21). Chinda et
al (22) reported that the
administration of vildagliptin (another DPP-4 inhibitor) prior to
LAD occlusion in a rat ischemia/reperfusion model, modulated
cardiac electrophysiology by normalizing the shortening of the
effective refractory period, decreasing the occurrence of premature
ventricular contractions and increasing the ventricular
fibrillation threshold during the ischemic period. A previous study
demonstrated that GLP-1 (10 nM) increased calcium transients and
sarcoplasmic reticular Ca2+ contents by regulating the
expression of calcium handling proteins. Specifically, GLP-1
decreased total phospholamban expression and ryanodine receptor
phosphorylation at S2814 in HL-1 cardiomyocytes (23). GLP-1 has been shown to exert direct
effects on atrial myocyte electrophysiology (23); however, no studies to date have
focused on the effects of exendin-4 on atrial electrophysiological
properties in a heart failure model, at least to the best of our
knowledge. Li et al (24)
demonstrated that atrial APDs were prolonged in heart failure, a
finding consistent with that observed herein. In the present study,
it was shown that exendin-4 treatment shortened APDs. Moreover, it
was reported that exendin-4 treatment shortened APDs, and decreased
atrial tachyarrhythmias inducibility in the rat model of MI-induced
heart failure. As chronic heart failure creates a substrate for AF
(24,25), the beneficial effects of exendin-4 on
heart failure-induced atrial arrhythmias may be partly responsible
for the improvements in cardiac function elicited by the drug.
Furthermore, the effects of exendin-4 on atrial arrhythmogenesis
were independent of glucose control in a previous study (4).
In dogs with heart failure, interstitial fibrosis,
slow conduction and increased conduction heterogeneity result in an
increased susceptibility to AF. The key atrial electrophysiology
alterations that occur in chronic heart failure may be the caused
by interstitial fibrosis-induced changes in atrial conduction
(25). Local atrial conduction
abnormalities interrupt cardiac electrical continuity, resulting in
unidirectional conduction slowing, which may promote reentry
maintenance (26,27). In the present study, slow conduction
and increased TAT dispersion were observed in the rat model of
MI-induced heart failure. Exendin-4 treatment increased conduction
velocity and decreased TAT dispersion, and these changes may
underlie the stabilizing effects of this treatment on atrial
electrophysiology.
Atrial fibrosis is a major cause of AF and a strong
predictor of the clinical outcomes of patients with heart failure.
Atrial fibrosis-induced conduction alterations have been shown to
be a major substrate for patients with AF (19). Additionally, atrial fibrosis favors
unidirectional conduction slowing or blockage, which leads to
reentry initiation and re-entry maintenance (28). Given that the results of several
studies have indicated that atrial fibrosis facilitates AF
(27,28), it is reasonable to hypothesize that
exendin-4 reduces atrial fibrillation susceptibility by attenuating
atrial fibrosis.
A previous study demonstrated that TGF-β1 functions
as a fibrogenic cytokine by stimulating collagen synthesis,
resulting in increased Col I and Col III expression (29). A previous study demonstrated that
chronic treatment with angiotensin II increased TGF-β expression
levels, a change associated with worse cardiac fibrosis and
enhanced cardiac collagen synthesis (30). In the present study, TGF-β1
expression was increased in the MI-induced heart failure model, and
exendin-4 attenuated TGF-β1 expression, a change accompanied by
decreases in Col I and Col III expression, suggesting that atrial
fibrosis is modulated by exendin-4. These results were consistent
with those of a previous study (31), in which the treatment of macrophages
with exendin-4 attenuated the increased expression of several
cytokines (α-smooth muscle actin, connective tissue growth factor
and TGF-β1) under high glucose conditions and TGF-β
stimulation.
The GLP-1 receptor has been reported to be located
in atrial myocytes and arteries; however, it is not expressed in
fibroblasts (8), which suggests that
the effects of exendin-4 on atrial fibrosis may occur secondary to
paracrine signaling. A previous study demonstrated that exendin-4
had no effect on the TGF-β-induced differentiation of cardiac
fibroblasts, indicating that the effects of exendin-4 on atrial
fibrosis may be mediated by another cell type (32). Tate et al (31) reported that exendin-4 exerted an
effect on paracrine communication between macrophages and cardiac
fibroblasts, which may provide an explanation for the observation
of the present study. It was also demonstrated that GLP-1 receptor
expression levels were decreased in atrial tissues following MI and
were normalized by exendin-4 treatment (31).
A previous study focusing on the acute
cardioprotective effects of exendin-4 in a rat ischemia/reperfusion
model demonstrated that PI3K/AKT signaling pathway activation
exerted cardioprotective effects attributable to its pro-survival
and antiapoptotic effects (12).
However, chronic cardiomyocyte-specific AKT activation has been
shown to promote both pathological hypertrophy and fibrosis.
Transgenic mice with cardiac-specific AKT-overexpression exhibit
cardiac hypertrophy at the molecular and histological levels
(33). Moreover,
Akt1-/- mice have been shown to be
protected from hypoxia and TGF-β-induced lung fibrosis, while
Akt-overexpressing mice exhibit pulmonary vascular fibrosis
(34). The findings of
aforementioned studies support the hypothesis that
exendin-4-induced PI3K/AKT signaling pathway inactivation may
contribute to the effects of exendin-4 on atrial fibrosis. In the
present study, the role of the PI3K/AKT signaling pathway in the
development of atrial fibrosis was investigated and it was found
that rats with MI exhibited increased PI3K/AKT phosphorylation in
atrial tissue, and abnormalities that were attenuated by treatment
with exendin-4; however, these effects were partly abolished by
co-administration with exendin9-39. These data suggested that the
PI3K/AKT signaling pathway is the downstream target of the GLP-1
receptor in atrial tissues in the MI-induced heart failure
model.
There are several limitations to the present study.
Firstly, only the electrophysiology in LA responding to exendin-4
was assessed. As LA was the main substrate of AF, only the response
of LA to exendin-4 treatment was measured. Future studies should
explore the difference between RA and LA responding to extenin-4.
Atrial conduction and atrial fibrosis were aspects of atrial
arrhythmogenesis, as well as ionic current and calcium handling. In
future studies, the effects of exendin-4 on ionic current and
calcium handling should be explored.
In conclusion, the results of the present study
demonstrated that the GLP-1 receptor agonist, exendin-4, decreased
atrial fibrosis and susceptibility to atrial arrhythmogenesis in
the rat model of MI-induced heart failure. The beneficial effects
of exendin-4 were partially diminished by the co-administration of
exendin9-39. Moreover, it was shown that exendin-4 exerted its
antifibrotic effects partly by inhibiting the PI3K/AKT signaling
pathway via the GLP-1 receptor. These data provide new insight
regarding the association between atrial electrophysiology and
exendin-4.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 81960047), Science
and Technology Foundation of Guiyang City [grant no. (2019)-9-1],
the Science and Technology Foundation of Health commission of
Guizhou Province (grant no. gzwjkj2019-1-092), and the Science and
Technology Foundation of Guizhou Provence [grant no.
(2019)2800].
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
WL conceived, designed and supervised the present
study. JinC and SX performed the experiments, reviewed the
manuscript, and analyzed and interpreted the data. LoW, WZ, PL, ND,
QT, YL, LiW and JiuC performed the experiments. All authors read
and approved the fnal manuscript.
Ethics approval and consent to
participate
All experimental protocols were approved by the
Experimental Animal Care and Institutional Animal Ethical Committee
of Guizhou Medical University (Guiyang, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Kemp CD and Conte JV: The pathophysiology
of heart failure. Cardiovasc Pathol. 21:365–371. 2012.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Lau DH, Schotten U, Mahajan R, Antic NA,
Hatem SN, Pathak RK, Hendriks JM, Kalman JM and Sanders P: Novel
mechanisms in the pathogenesis of atrial fibrillation: Practical
applications. Eur Heart J. 37:1573–1581. 2016.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Verheule S, Eckstein J, Linz D, Maesen B,
Bidar E, Gharaviri A and Schotten U: Role of endo-epicardial
dissociation of electrical activity and transmural conduction in
the development of persistent atrial fibrillation. Prog Biophys Mol
Biol. 115:173–185. 2014.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Robinson E, Cassidy RS, Tate M, Zhao Y,
Lockhart S, Calderwood D, Church R, McGahon MK, Brazil DP,
McDermott BJ, et al: Exendin-4 protects against post-myocardial
infarction remodelling via specific actions on inflammation and the
extracellular matrix. Basic Res Cardiol. 110(20)2015.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Noyan-Ashraf MH, Momen MA, Ban K, Sadi AM,
Zhou YQ, Riazi AM, Baggio LL, Henkelman RM, Husain M and Drucker
DJ: GLP-1R agonist liraglutide activates cytoprotective pathways
and improves outcomes after experimental myocardial infarction in
mice. Diabetes. 58:975–983. 2009.PubMed/NCBI View Article : Google Scholar
|
|
6
|
DeNicola M, Du J, Wang Z, Yano N, Zhang L,
Wang Y, Qin G, Zhuang S and Zhao TC: Stimulation of glucagon-like
peptide-1 receptor through exendin-4 preserves myocardial
performance and prevents cardiac remodeling in infarcted
myocardium. Am J Physiol Endocrinol Metab. 307:E630–E643.
2014.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Bao W, Aravindhan K, Alsaid H, Chendrimada
T, Szapacs M, Citerone DR, Harpel MR, Willette RN, Lepore JJ and
Jucker BM: Albiglutide, a long lasting glucagon-like peptide-1
analog, protects the rat heart against ischemia/reperfusion injury:
Evidence for improving cardiac metabolic efficiency. PLoS One.
6(e23570)2011.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Richards P, Parker HE, Adriaenssens AE,
Hodgson JM, Cork SC, Trapp S, Gribble FM and Reimann F:
Identification and characterization of GLP-1 receptor-expressing
cells using a new transgenic mouse model. Diabetes. 63:1224–1233.
2014.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Chen J, Wang D, Wang F, Shi S, Chen Y,
Yang B, Tang Y and Huang C: Exendin-4 inhibits structural
remodeling and improves Ca2+ homeostasis in rats with heart failure
via the GLP-1 receptor through the eNOS/cGMP/PKG pathway. Peptides.
90:69–77. 2017.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Iwasa M, Kobayashi H, Yasuda S, Kawamura
I, Sumi S, Yamada Y, Shiraki T, Yamaki T, Ushikoshi H, Aoyama T, et
al: Antidiabetic drug voglibose is protective against
ischemia-reperfusion injury through glucagon-like peptide 1
receptors and the phosphoinositide 3-kinase-Akt-endothelial nitric
oxide synthase pathway in rabbits. J Cardiovasc Pharmacol.
55:625–634. 2010.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Wende AR, O'Neill BT, Bugger H, Riehle C,
Tuinei J, Buchanan J, Tsushima K, Wang L, Caro P, Guo A, et al:
Enhanced cardiac Akt/protein kinase B signaling contributes to
pathological cardiac hypertrophy in part by impairing mitochondrial
function via transcriptional repression of mitochondrion-targeted
nuclear genes. Mol Cell Biol. 35:831–846. 2015.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Basalay MV, Mastitskaya S, Mrochek A,
Ackland GL, Del AA, Sanchez J, Sjoquist PO, Pernow J, Gourine AV
and Gourine A: Glucagon-like peptide-1 (GLP-1) mediates
cardioprotection by remote ischaemic conditioning. Cardiovasc Res.
112:669–676. 2016.PubMed/NCBI View Article : Google Scholar
|
|
13
|
National Research Council (US) Committee
for the Update of the Guide for the Care and Use of Laboratory
Animals. Guide for the Care and Use of Laboratory Animals. 8th
edition. Washington (DC): National Academies Press (US), 2011.
|
|
14
|
Yamamoto H, Lee CE, Marcus JN, Williams
TD, Overton JM, Lopez ME, Hollenberg AN, Baggio L, Saper CB,
Drucker DJ and Elmquist JK: Glucagon-like peptide-1 receptor
stimulation increases blood pressure and heart rate and activates
autonomic regulatory neurons. J Clin Invest. 110:43–52.
2002.PubMed/NCBI View
Article : Google Scholar
|
|
15
|
Sisley S, Smith K, Sandoval DA and Seeley
RJ: Differences in acute anorectic effects of long-acting GLP-1
receptor agonists in rats. Peptides. 58:1–6. 2014.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Wang D, Liu T, Shi S, Li R, Shan Y, Huang
Y, Hu D and Huang C: Chronic administration of catestatin improves
autonomic function and exerts cardioprotective effects in
myocardial infarction rats. J Cardiovasc Pharmacol Ther.
21:526–535. 2016.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Amino M, Yoshioka K, Tanabe T, Tanaka E,
Mori H, Furusawa Y, Zareba W, Yamazaki M, Nakagawa H, Honjo H, et
al: Heavy ion radiation up-regulates Cx43 and ameliorates
arrhythmogenic substrates in hearts after myocardial infarction.
Cardiovasc Res. 72:412–421. 2006.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Nattel S: Molecular and cellular
mechanisms of atrial fibrosis in atrial Fibrillation. JACC Clin
Electrophysiol. 3:425–435. 2017.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Chen J, Xu S, Zhou W, Wu L, Wang L and Li
W: Exendin-4 reduces ventricular arrhythmia activity and calcium
sparks-mediated sarcoplasmic reticulum Ca Leak in rats with heart
failure. Int Heart J. 61:145–152. 2020.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Lee TM, Chen WT and Chang NC: Sitagliptin
decreases ventricular arrhythmias by attenuated glucose-dependent
insulinotropic polypeptide (GIP)-dependent resistin signalling in
infarcted rats. Biosci Rep. 36(e00307)2016.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Chinda K, Palee S, Surinkaew S,
Phornphutkul M, Chattipakorn S and Chattipakorn N: Cardioprotective
effect of dipeptidyl peptidase-4 inhibitor during
ischemia-reperfusion injury. Int J Cardiol. 167:451–457.
2013.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Huang JH, Chen YC, Lee TI, Kao YH, Chazo
TF, Chen SA and Chen YJ: Glucagon-like peptide-1 regulates calcium
homeostasis and electrophysiological activities of HL-1
cardiomyocytes. Peptides. 78:91–98. 2016.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Li D, Melnyk P, Feng J, Wang Z, Petrecca
K, Shrier A and Nattel S: Effects of experimental heart failure on
atrial cellular and ionic electrophysiology. Circulation.
101:2631–2638.22. 2000.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Li D, Fareh S, Leung TK and Nattel S:
Promotion of atrial fibrillation by heart failure in dogs: Atrial
remodeling of a different sort. Circulation. 100:87–95.
1999.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Lammers WJ, Schalij MJ, Kirchhof CJ and
Allessie MA: Quantification of spatial inhomogeneity in conduction
and initiation of reentrant atrial arrhythmias. Am J Physiol.
259:H1254–H1263. 1990.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Jalife J and Kaur K: Atrial remodeling,
fibrosis, and atrial fibrillation. Trends Cardiovasc Med.
25:475–484. 2015.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Vigmond E, Pashaei A, Amraoui S, Cochet H
and Hassaguerre M: Percolation as a mechanism to explain atrial
fractionated electrograms and reentry in a fibrosis model based on
imaging data. Heart Rhythm. 13:1536–1543. 2016.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Purnomo Y, Piccart Y, Coenen T, Prihadi JS
and Lijnen PJ: Oxidative stress and transforming growth
factor-β1-induced cardiac fibrosis. Cardiovasc Hematol Disord Drug
Targets. 13:165–172. 2013.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Zhao W, Zhao T, Chen Y, Ahokas RA and Sun
Y: Oxidative stress mediates cardiac fibrosis by enhancing
transforming growth factor-beta1 in hypertensive rats. Mol Cell
Biochem. 317:43–50. 2008.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Tate M, Robinson E, Green BD, McDermott BJ
and Grieve DJ: Exendin-4 attenuates adverse cardiac remodelling in
streptozocin-induced diabetes via specific actions on infiltrating
macrophages. Basic Res Cardiol. 111(1)2016.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Ban K, Noyan-Ashraf MH, Hoefer J, Bolz SS,
Drucker DJ and Husain M: Cardioprotective and vasodilatory actions
of glucagon-like peptide 1 receptor are mediated through both
glucagon-like peptide 1 receptor-dependent and -independent
pathways. Circulation. 117:2340–2350. 2008.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Condorelli G, Drusco A, Stassi G,
Bellacosa A, Roncarati R, Iaccarino G, Russo MA, Gu Y, Dalton N,
Chung C, et al: Akt induces enhanced myocardial contractility and
cell size in vivo in transgenic mice. Proc Natl Acad Sci USA.
99:12333–12338. 2002.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Abdalla M, Sabbineni H, Prakash R, Ergul
A, Fagan SC and Somanath PR: The Akt inhibitor, triciribine,
ameliorates chronic hypoxia-induced vascular pruning and
TGFβ-induced pulmonary fibrosis. Br J Pharmacol. 172:4173–4188.
2015.PubMed/NCBI View Article : Google Scholar
|















