Introduction
Primary carnitine deficiency (PCD) is a fatal
autosomal recessive disorder of fatty acid oxidation resulting from
the defective transportation of carnitine into the kidney, heart
and muscles (1). The SLC22A5 gene
was originally identified in these tissues (1). PCD is typically characterized by
episodes of hypoketotic hypoglycemia, hepatomegaly, elevated
transaminases and hyperammonemia in infants; skeletal myopathy,
high levels of creatine kinase and cardiomyopathy in childhood; or
fatigability in adulthood (2-4).
Case report
A 15-month-old girl was admitted to Zhongshan
Hospital Affiliated to Sun Yat-Sen University (Zhongshan, China) in
November 2015 due to cough that had lasted for 3 days and a
seizure, which lasted for 5 h. She did not present wheezing,
cyanosis, fever or vomiting and exhibited no notable symptoms. No
abnormalities were noted at birth, with a birth weight of 3.1 kg
and a body length of 50 cm. The physical and motor development was
normal. The patient exhibited recurrent forced clonic convulsions
4-5 times/day following admission and suffered from a coma with
convulsions. No abnormalities were observed in the cardiopulmonary
and abdominal physical examination, and physiological reflexes were
present with Babinski sign. The white blood cell count in the
peripheral blood was 16,160 cells/mm3. The results of
serum c-reactive protein, lactic acid, blood ammonia biochemistry
and cerebrospinal fluid analyses were normal. The results of an
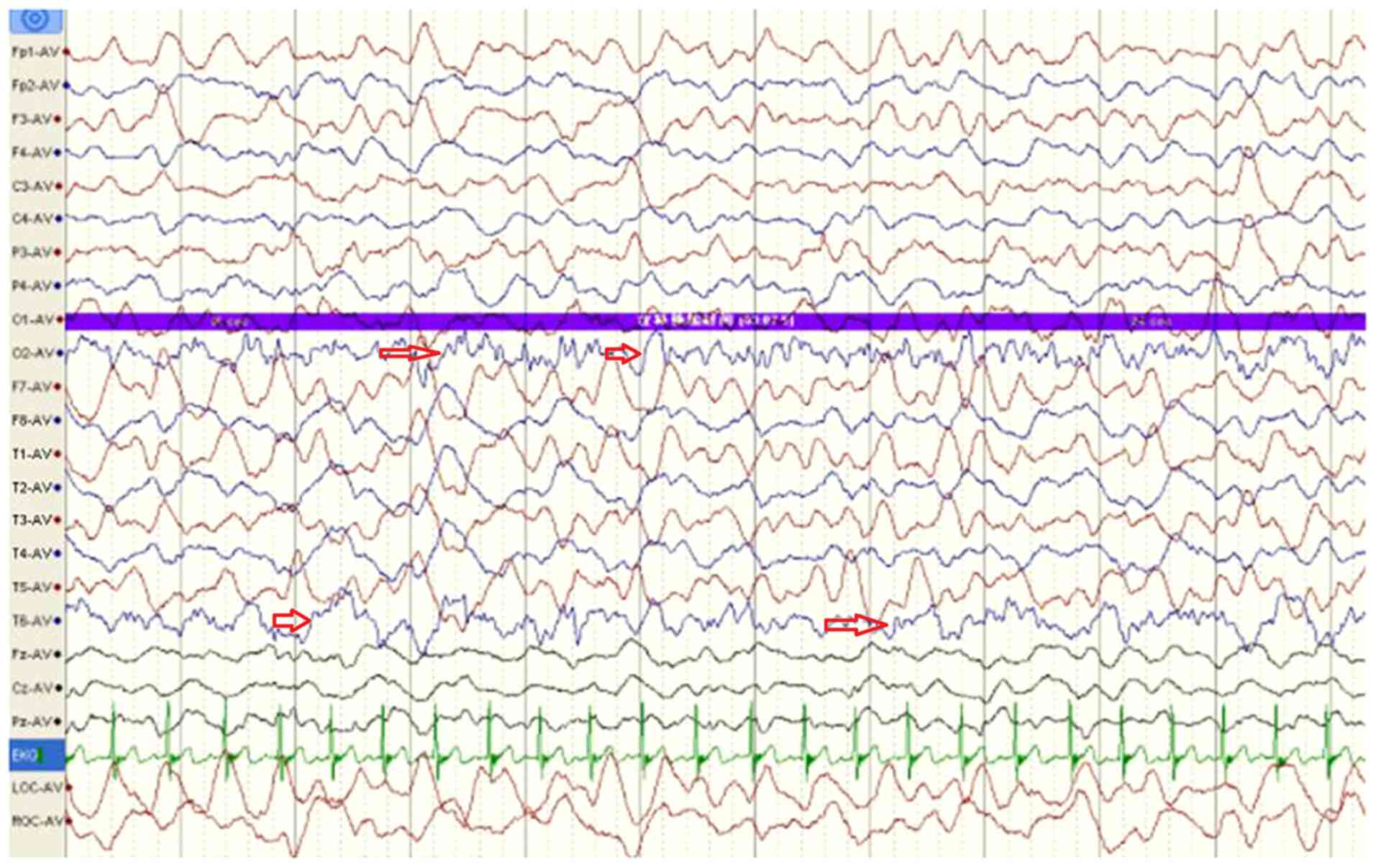
electroencephalogram indicated large and slow waves originating
from multiple right occipital areas and large paroxysmal wave
rhythms in the left occipital area (Fig. 1; arrows). Echocardiography revealed
a normal internal structure and heart functions. Brain MRI
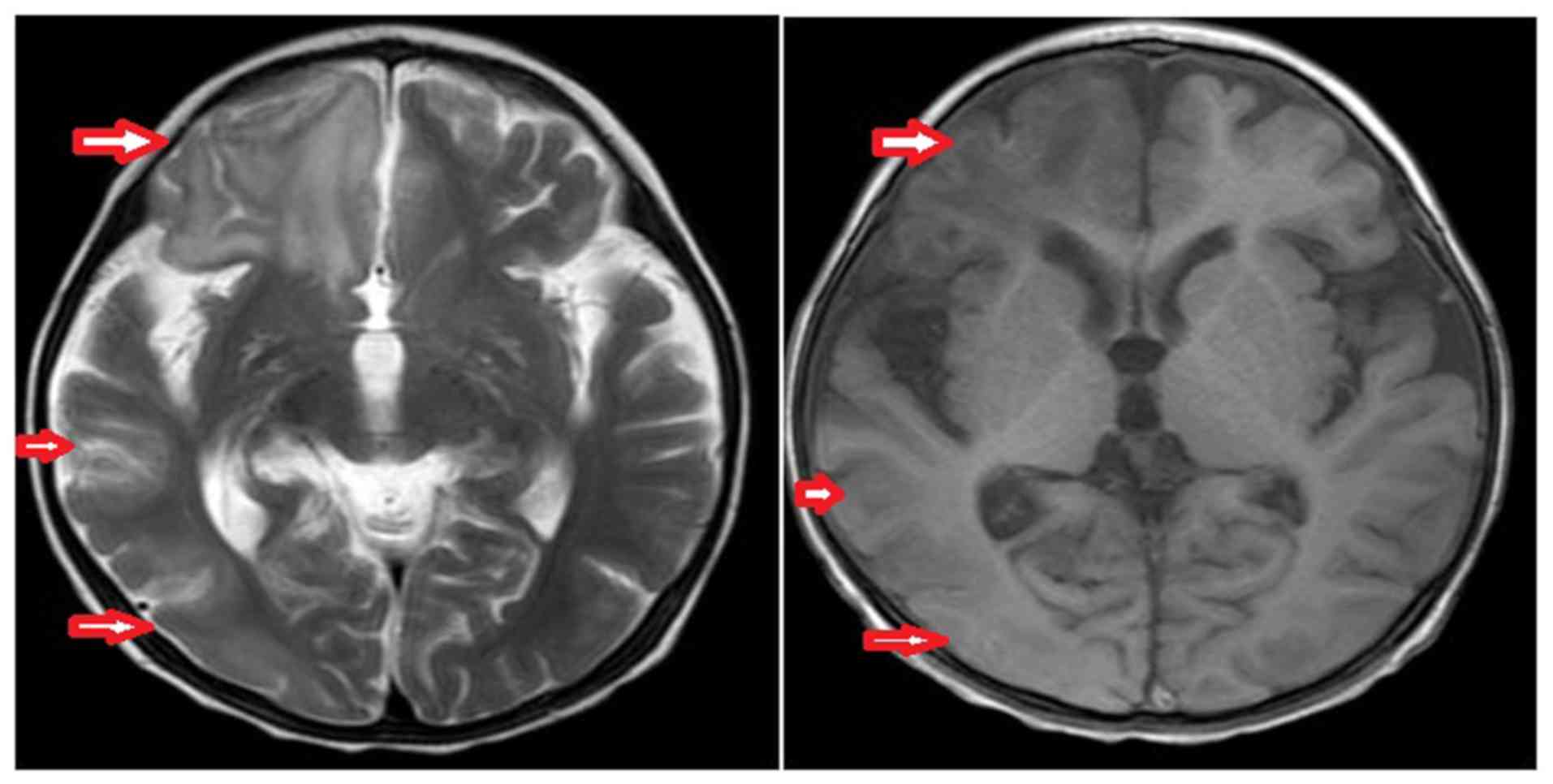
(November 2015) indicated a low signal in the right cerebral
hemisphere on the T1-weighted image and a high signal on the
T2-weighted image, which were primarily localized in the parietal
occipital temporal lobe. The ventricular system was symmetrical
with no enlargement, and the midline did not shift (Fig. 2). The disease management included
treatment with ceftriaxone (100 mg/kg/day), immunoglobulin (1
g/kg/day for 2 days), methylprednisolone (2 mg/kg/day), mannitol (1
g/kg/day) and airway nursing, as well as a symptomatic treatment
with oxcarbazepine (15 mg/kg; twice/day) as an antiepileptic drug.
The screening for metabolic diseases revealed that plasma free
carnitine level was low (1.3 µmol/l; normal range, 25-50 µmol/l),
indicating that the patient suffered from PCD. The patient was
treated with levocarnitine at a dose of 50-400 mg/kg/day divided
into three doses and a low-fat and medium-chain triglyceride-rich
diet. The plasma free carnitine levels of the patient returned to
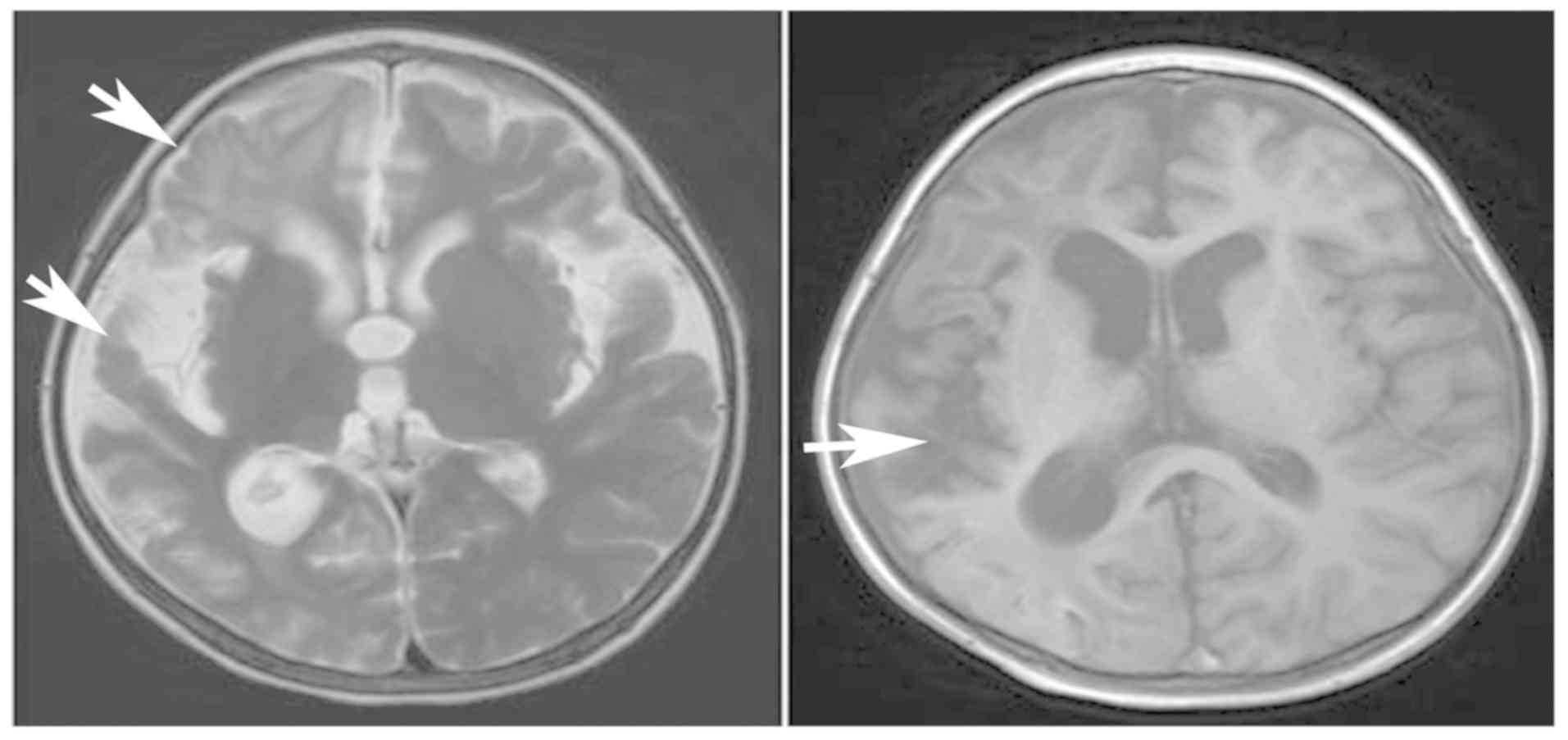
normal levels after 1 month. Brain MRI (February 2016) indicated
atrophy in the right cerebral hemisphere and an enlarged cortical
lamellar necrosis (Fig. 3). The
patient suffered from intractable epilepsy despite oral
oxcarbazepine administration for 8 months. Subsequently, the
antiepileptic therapy was altered to oral sodium valproate (5 ml;
twice/day) and oral levetiracetam (0.375 g; twice/day). Despite
these treatments, the epileptic symptoms remained recurrent. In
order to control the epilepsy, the patient was subjected to a right
cerebral resection to excise the severely diseased lateral brain
area in June 2017, following which the epilepsy was controlled.
Brain MRI scans (August 2017) demonstrated alterations following
right cerebral hemisphere resection and right cerebral basal
ganglia resection, with compensatory hydrocephalus in the right
frontotemporal occipital lobe and chronic hematoma and local
crescent subdural effusion in the left frontal lobe (Fig. 4). The muscle strength and tension of
the right limbs and the left upper limb of the patient were normal,
but the muscle power of the left lower limb was grade 4 according
to the grading system of muscle power (5), and the muscle tone was determined as
paratonia of hypertonia (5). In
addition, the language ability of the patient was normal.
The older sister of the proband, aged 3 years and 10
months, complained of malaise and fatigue that lasted for 1 week,
and was admitted to Zhongshan Hospital Affiliated to Sun Yat-Sen
University in February 2016. She did not present with cough, chest
tightness, shortness of breath, cyanosis, vomiting, fever, diarrhea
or convulsions. Her appetite and urine were normal. The physical
and auxiliary examinations indicated that she exhibited PCD with
cardiomyopathy complications, but without damage to the other
organs. Echocardiography indicated a left atrial and left
ventricular enlargement, as well as cardiac insufficiency. The left
atrial diameter was 24 mm, and the left ventricular diameter was 40
mm. In addition, the left ventricular ejection fraction was 48%.
Brain MRI presented no abnormalities. The blood amino acid and
acylcarnitine spectrum analysis of inherited metabolic diseases
indicated that free carnitine (0.66 µmol/l; normal range, 25-50
µmol/l) and a variety of acylcarnitine compounds (e.g.
succinylcarnitine, butyrylcarnitine and propionylcarnitine, among
others) were low, suggesting PCD. She was immediately treated with
levocarnitine (100 mg/kg daily) and a low-fat and rich-medium-chain
triglyceride diet, and the symptoms improved after 3 months. The
size and function of her heart were restored to normal with the
left atrial diameter of 21 mm, the left ventricular diameter of 35
mm, the left ventricular ejection fraction of 75% and normal
physical development following 3 months of therapy.
Four members of the family were screened for
congenital genetic metabolic diseases and SLC22A5 gene mutations
using high-performance (HP) liquid chromatography (LC) tandem mass
spectrometry (HPLC/MS/MS) and Sanger sequencing. For the HPLC, the
following systems and conditions were used: HPLC system (LC20ADXR;
Shimadzu Corporation); column, Agilent Zorbax SB-C18 3.5 µm,
2.1ⅹ100 mm (Agilent Technologies, Inc.); environment temperature,
25±2˚C; environment humidity, 35-80%; sample quantity (15 µl);
composition of mobile phase, one solvent of formic-and-methanol
solution from NeoBase Non-derivatized MSMS kit (PerkinElmer, Inc.);
and flow rate, 0.235 ml/min for sample injection before 0.20 min;
0.017 ml/min for ionization from 0.21 to 1.10 min; 0.7 ml/min for
flushing the pipe from 1.15 to 1.45 min; and 0.235 ml/min for
equilibrium to the end. For the LC/MS/MS, in order to improve the
sensitivity, the analytical measurements were performed in the
multiple reaction monitoring mode (MRM). The MRM parameters were
set according to the operating manual provided by Neobase
Non-derivatized MSMS kit (MRM transition masses, CO 162.20 m/z
>103.10 m/z, CO IS 171.20 m/z >103.10 m/z, C2 241.10 m/z
>85.10 m/z, C2 IS 207.10 m/z >85.10 m/z, C3 218.10 m/z
>85.10 m/z, C3 IS 221.10 m/z >85.10 m/z; Dwell time, 0.025
sec; collision energy, 14-18 V). The following systems and
conditions were used: LC/MS/MS system (AB SCIEX API 3200 MD; AB
Sciex LLC); ionization mode, positive; nitrogen gas temperature,
400˚C, nebuliser pressure, 30 psi; and flow rate, 0.235 ml/min. For
Sanger sequencing, after informed consent of the subjects'
guardians, 2 ml venous blood (with EDTA anticoagulation) were
collected from the four members of the family. Genomic DNA was
extracted via routine phenol chlorine method. The primers were
designed with Primer Premier version 5.0 software (Premier Biosoft
International) to amplify all exons and introns of the SLC22A5
gene. A PCR machine (C1000; Bio-Rad Laboratories, Inc.) was used to
amplify the extracted DNA. The PCR reaction volume was 25 µl,
including Takara La Taq premix (Takara Biotechnology Co., Ltd.)
12.5 µl, upstream and downstream primer mixture 0.75 µl (10
pmol/µl), genomic DNA 100 ng and deionized water to 25 µl. The PCR
reaction conditions were as follows: Pre-denaturation at 95˚C for 3
min; denaturation at 94˚C for 30 sec, annealing at 60˚C for 30 sec,
extension for 40 sec at 72˚C for 38 cycles; and final extension for
8 min at 72˚C. The amplified PCR products were identified by 1.5%
agarose gel electrophoresis (Bio-Rad Laboratories, Inc.), and the
sequencing results were compared with the SLC22A5 gene sequences of
the human genome in GenBank (http://www.ncbi.nlm.nih.gov/Genbank). One nonsense
mutation [c.760C>T (p.R254X)] was identified. The two sisters
exhibited homozygous mutations, whereas their parents exhibited
heterozygous mutations (Fig. 5).
The plasma free carnitine levels of the parents were normal.
Electrocardiogram, ultrasound, biochemical and brain MRI
examinations of the family members were performed; the results
demonstrated that all family members had not developed episodes of
hypoketotic hypoglycemia, hepatomegaly, elevated transaminases or
hyperammonemia. The parents were asymptomatic.
Discussion
PCD is an autosomal recessive disease caused by a
functional defect in the carnitine transporter, which is encoded by
the SLC22A5 gene (6). The incidence
rate of PCD in Japan and Europe is 1:40,000-1:120,000 (7,8). The
principal function of levocarnitine is to assist in the transport
of long chain fatty acids into the mitochondria to undergo
β-oxidation (9). High-performance
liquid chromatography tandem mass spectrometry (HPLC/MS/MS) is one
of the methods that are used to test for metabolic disorders
(10,11); therefore, HPLC/MS/MS screening was
applied in the present case report. Carnitine deficiency has been
indicated to result in a reduction of energy generation and the
accumulation of long chain fatty acids in the cytoplasm, as well as
a number of biochemical abnormalities (hypoglycemia, liver
dysfunction, etc.) and organ damage (12,13).
The principal clinical manifestations include hypoketotic
hypoglycemia, encephalopathy, cardiomyopathy and skeletal muscle
weakness. The diagnoses of PCD in the present case report were
based on the low levels of plasma free carnitine (<5 µmol/l).
Lin et al (14) investigated
the potential mutations in the SLC22A5 gene in patients with PCD,
and the diagnosis of eight patients with PCD was confirmed at the
gene level. A total of six mutations were identified in the SLC22A5
gene, including one novel mutation, which expanded the existing
mutation spectrum of the SLC22A5 gene (14). In the general population, the
incidence of heterozygous mutations ranges between 0.5 and 1%, and
most frequently occurs between 1 month and 7 years of age (7). The SLC22A5 gene is localized in
chromosome 5q31 and contains 10 exons (15). The reported mutations are localized
in exons 1-9 and introns 3, 7 and 8, and exonal mutations have been
indicated to be the most frequent (16). The low levels of plasma free
carnitine are an indirect effect of the dysregulation in glucose
aerobic oxidation, gluconeogenesis and ketogenesis, as well as
other metabolic pathways, which has been reported to result in
metabolic disorders and organ damage (15). When carnitine concentrations in the
plasma are 10-20% lower than normal levels, carnitine deficiencies
may induce a number of clinical symptoms in the liver, heart,
muscles and brain (15). In 1975,
the first case if PCD was reported, since which the disease has
received increasing clinical attention (17). Over the last decade, owing to the
use of MS/MS analysis of dried blood spots, the acylcarnitine
spectrum and improvements in the determination of amino acid
chromatography and urine organic acid gas mass spectrometry for
genetic metabolic disease detection, a greater number of children
have received timely diagnosis (18,19).
The therapeutic dose of levocarnitine should be
adjusted according to alterations in its concentration in the blood
of individual patients, as well as the blood alkalinity and the
degree of the disease in order to maintain the stability of the
tissues and blood. A high dose levocarnitine treatment may cause
diarrhea, nausea and other gastrointestinal discomforts; however,
the dose can be reduced and once the adverse reactions have
improved, it can be gradually increased to the initial treatment
dose (20). Patients with PCD
require a lifelong treatment with levocarnitine, and a sudden
withdrawal may result in a rapid drop in the plasma carnitine
concentration, causing recurrent Reye syndrome and even sudden
death (21). For asymptomatic
patients with PCD, treatment with levocarnitine has been indicated
to be effective in preventing morbidity and sudden death (22). In the present case report, although
the two sisters with PCD exhibited a homozygous mutation in SLC22A5
[c.760 C>T (p.R254X)], their clinical manifestations varied.
Following treatment of the proband with levocarnitine and internal
medical therapies (including antiepileptic therapy and
rehabilitation), the serum free carnitine levels were restored to
normal levels, but the clinical symptoms recovered slowly, and
sequelae in the nervous system remained. A 3-year pilot study in
the Zhejiang province in China has suggested that mental
retardation and motor delay were difficult to reverse in the
majority of symptomatic patients with PCD following a late
diagnosis (23). In the present
study, although the older sister of the proband exhibited cardiac
complications, following oral administration of levocarnitine, the
clinical symptoms improved rapidly. It has been reported that oral
carnitine supplementation in infants with PCD-related
cardiomyopathy result in improved clinical outcomes with the
ejection fraction reaching 75% following 3-month treatment
(24,25). Previous reports have also suggested
that excising the severely diseased lateral brain may be used to
treat refractory epilepsy, with epilepsy and functional outcomes
being improved following surgery (26-28),
which was in agreement with the treatment results observed in the
proband in the present case report.
In conclusion, the two cases and the pedigree
analysis revealed that clinical manifestations of PCD within the
same family were specific to each individual. The results also
demonstrated that treatment with levocarnitine supplementation
should be initiated as early as possible before irreversible organ
damage occurs. In addition, metabolic decompensation and cardiac
muscle functions were demonstrated to improve after carnitine
supplementation. The resection of the severely diseased unilateral
brain combined with carnitine supplementation and antiepileptic
therapy may be an effective treatment strategy for PCD with
intractable epilepsy.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
XFY conceived the study, participated in its design
and coordination and drafted the manuscript. GSL performed the
clinical diagnosis and treatment of the patients. BY performed the
molecular genetic studies, participated in the sequence alignment
and drafted the manuscript. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Informed consent for participation was received from
the patients' guardians, and the protocol was approved by the
Ethical Committee of Zhongshan Hospital Affiliated to Sun Yat-Sen
University, Zhongshan, China (approval no. B2015111601).
Patient consent for publication
Informed consent was obtained from the guardians of
the two patients.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Tan JQ, Chen DY, Li ZT, Yan TZ, Huang JW
and Cai R: Genetic diagnosis of 10 neonates with primary carnitine
deficiency. Zhongguo Dang Dai Er Ke Za Zhi. 19:1150–1154.
2017.PubMed/NCBI View Article : Google Scholar : (In Chinese).
|
|
2
|
Buist NR: Historical perspective on
clinical trials of carnitine in children and adults. Ann Nutr
Metab. 68:1–4. 2016.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Ravindranath A, Pai G, Srivastava A,
Poddar U and Yachha SK: Infant with hepatomegaly and hypoglycemia:
A setting for fatty acid oxidation defects. Indian J Gastroenterol.
36:429–434. 2017.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Belousova ED: The decreased level of
plasma carnitine in patients with epilepsy. Zh Nevrol Psikhiatr Im
S S Korsakova. 117:106–110. 2017.PubMed/NCBI View Article : Google Scholar : (In Russian).
|
|
5
|
Greenberg DA, Aminoff MJ and Simon RP:
Clinical Neurogy (5th edition). The United States: Mc Graw-Hill,
New York, NY, pp157-158, 2002.
|
|
6
|
Ferdinandusse S, Brinke HT, Ruiter JP,
Haasjes J, Oostheim W, Ven Lenthe H, IJlst L, Ebberink MS, Wanders
RJ, Vaz FM and Waterham HR: Mutation creating an upstream
translation initiation codon in SLC22A5 5'UTR is a frequent cause
of primary carnitine deficiency. Hum Mutat. 40:1899–1904.
2019.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Koizumi A, Nozaki J, Ohura T, Kayo T, Wada
Y, Nezu J, Ohashi R, Tamai I, Shoji Y, Takada G, et al: Genetic
epidemiology of the carnitine transporter OCTN2 gene in a Japanese
population and phenotypic characterization in Japanese pedigrees
with primary systemic carnitine deficiency. Hum Mol Genet.
8:2247–2254. 1999.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Hassan FA, El Mougy F, Sharaf SA, Mandour
I, Morgan MF, Selim LA, Hassan SA, Salem F, Oraby A, Girgis MY, et
al: Inborn errors of metabolism detectable by tandem mass
spectrometry in Egypt: The first newborn screening pilot study. J
Med Screen. 23:124–129. 2016.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Nałęcz KA and Nałęcz MJ:
Carnitine-mitochondria and beyond. Postepy Biochem. 62:85–93.
2016.PubMed/NCBI
|
|
10
|
Lindner M, Gramer G, Haege G,
Fang-Hoffmann J, Schwab KO, Tacke U, Trefz FK, Mengel E, Wendel U,
Leichsenring M, et al: Efficacy and outcome of expanded newborn
screening for metabolic diseases-report of 10 years from South-West
Germany. Orphanet J Rare Dis. 6(44)2011.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Lehotay DC, Hall P, Lepage J, Eichhorst
JC, Etter ML and Greenberg CR: LC-MS/MS progress in newborn
screening. Clin Biochem. 44:21–31. 2011.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Longo N, Frigeni M and Pasquali M:
Carntiin transport and fatty acid oxidation. Biochim Biophys Acta.
1863:2422–2435. 2016.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Gallant NM, Leydiker K, Wilnai Y, Lee C,
Lorey F, Feuchtbaum L, Tang H, Carter J, Enns GM, Packman S, et al:
Biochemical characteristics of newborns with carnitine transporter
defect identified by newborn screening in California. Mol Genet
Metab. 122:76–84. 2017.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Lin Y, Lin W, Yu K, Zheng F, Zheng Z and
Fu Q: Mutational analysis of SLC22A5 gene in eight patients with
systemic primary carnitine deficiency. Zhonghua Yi Xue Yi Chuan Xue
Za Zhi. 34:35–39. 2017.PubMed/NCBI View Article : Google Scholar : (In Chinese).
|
|
15
|
Frigeni M, Balakrishnan B, Yin X, Calderon
FR, Mao R, Pasquali M and Longo N: Functional and molecular studies
in primary carnitine deficiency. Hum Mutat. 38:1684–1699.
2017.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Li FY, El-Hattab AW, Bawle EV, Boles RG,
Schmitt ES, Scaglia F and Wong LJ: Molecular spectrum of SLC22A5
(OCTN2) gene mutations detected in 143 subjects evaluated for
systemic carnitine defciency. Hum Mutat. 31:E1632–E1651.
2010.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Angelini C: Letter: Carnitine Deficiency.
Lancet. 2(554)1975.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Longo N: Primary carnitine deficiency and
newborn screening for disorders of the carnitinecycle. Ann Nutr
Metab. 68:5–9. 2016.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Zheng J, Zhang Y, Hong F, Yang J, Tong F,
Mao H and Huang X, Zhou X, Yang R, Zhao Z and Huang X: Screening
for fatty acid oxidation disorders of newborns in Zhejiang
province: prevalence, outcome and follow-up. Zhejiang Da Xue Xue
Bao Yi Xue Ban. 46:248–255. 2017.PubMed/NCBI(In Chinese).
|
|
20
|
Deswal S, Bijarnia-Mahay S, Manocha V,
Hara K, Shigematsu Y, Saxena R and Verma IC: Primary carnitine
deficiency-a rare treatable cause of cardiomyopathy and massive
hepatomegaly. Indian J Pediatr. 84:83–85. 2017.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Lahrouchi N, Lodder EM, Mansouri M, Tadros
R, Zniber L, Adadi N, Clur SA, van Spaendonck-Zwarts KY, Postma AV,
Sefiani A, et al: Exome sequencing identifies primary carnitine
deficiency in a family with cardiomyopathy and sudden death. Eur J
Hum Genet. 25:783–787. 2017.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Madsen KL, Preisler N, Rasmussen J,
Hedermann G, Olesen JH, Lund AM and Vissing J: L-Carnitine improves
skeletal muscle fat oxidation in primary carnitine deficiency. J
Clin Endocrinol Metab. 103:4580–4588. 2018.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Jun JS, Lee EJ, Park HD and Kim HS:
Systemic primary carnitine deficiency with hypoglycemic
encephalopathy. Ann Pediatr Endocrinol Metab. 21:226–229.
2016.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Perin F, Rodríguez-Vázquez Del Rey MD,
Carreras-Blesa C, Arrabal-Fernández L, Jiménez-Jáimez J and
Tercedor L: Dilated cardiomyopathy with short QT interval suggests
primary carnitine deficiency. Rev Esp Cardiol (Engl Ed).
71:1074–1075. 2018.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Papadopoulou-Legbelou K, Gogou M, Dokousli
V, Eboriadou M and Evangeliou A: Dilated cardiomyopathy as the only
clinical manifestation of carnitine transporter deficiency. Indian
J Pediatr. 84:231–233. 2017.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Morrison-Levy N, Go C, Ochi A, Otsubo H,
Drake J, Rutka J and Weiss SK: Children with autism spectrum
disorders and drug-resistant epilepsy can benefit from epilepsy
surgery. Epilepsy Behav. 85:200–204. 2018.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Hirsch E and Arzimanoglou A: Children with
drug-resistant partial epilepsy: Criteria for the identification of
surgical candidates. Rev Neurol (Paris). 160:5S210–5S219.
2004.PubMed/NCBI(In French).
|
|
28
|
Roth J, Nagar S, Constantini S and Fried
I: Hemispherotomy for treatment of refractory epilepsy in children.
Harefuah. 156:482–485. 2017.PubMed/NCBI(In Hebrew).
|