Introduction
Inflammatory diseases, including infection,
endotoxemia and sepsis, are a significant burden on perioperative
management of patients (1).
Inflammation-induced organ dysfunction and failure can result in
severe multiple organ dysfunction syndromes (2). The central pathophysiology of
inflammation-induced organ dysfunction is the production of an
inflammatory cytokine storm caused by uncontrolled and unregulated
innate immune responses (3,4). Macrophages produce the majority of
cytokines, including IL-6, TNF-α and IL-1β, during inflammatory
diseases (3,4). Suppressing cytokine production by
macrophages can attenuate disease progression and improve outcomes
(5). However, the regulation and
associated mechanism of cytokine production in macrophages remain
largely unknown. The discovery of specific molecules or mechanisms
that support macrophage production may reveal potential targets for
the management of inflammation.
Previous studies have demonstrated that endoplasmic
reticulum (ER) stress serves an essential role in inflammation and
cytokine production by macrophages (6,7). The
activation of macrophages by lipopolysaccharides (LPS) leads to the
unfolded protein response, which results in the activation of three
ER stress markers on the ER membrane: Inositol-requiring enzyme 1α
(IRE1α), PKR-like endoplasmic reticulum kinase (PERK) and
activating transcription factor 6 (ATF6) (6). These three molecules regulate
subsequent signaling to NF-κB, Bcl-2 and nuclear factor erythroid 2
(Nrf2), regulating inflammation, oxidative stress and apoptosis
(8,9). Therefore, the activation of ER stress
is essential for the inflammatory response of macrophages.
Homeodomain-interacting protein kinase 2 (HIPK2) is
a serine/threonine kinase located in the nucleus (10). A previous study has reported that
HIPK2 functions as a tumor suppressor and that it promotes
apoptosis in response to chemotherapeutic drugs and radiation
(10). Furthermore, another
previous study proposed that HIPK2 sustains genomic stability by
enhancing DNA damage repair signaling (11). Apart from the role of HIPK2 that has
been elucidated within oncological research, a recent study
demonstrated that HIPK2 deficiency leads to the impaired production
of type I interferon in macrophages during antiviral immunity
(12). However, the function and
mechanism of HIPK2 in anti-microbial immunity remain poorly
investigated.
The present study investigated the function of HIPK2
in LPS-stimulated macrophages and the underlying mechanism of
cytokine production. Furthermore, the expression levels of HIPK2,
effects of HIPK2 knockdown and the crucial mechanism of its
regulation of cytokines in LPS-stimulated macrophages were
investigated.
Materials and methods
Animals and models
A total of 60 C57BL/6 male mice (weight, 20-25 g;
age, 6-8 weeks) were purchased from Shanghai SLAC Laboratory Animal
Co., Ltd. All mice were housed at a temperature of 18-22˚C with a
relative humidity of 50-60% and 12-h light-dark cycles, with free
access to food and water. The Ethics Committee of Changhai Hospital
(Shanghai, China) approved the animal experiments. For the murine
LPS-challenge model, each mouse was intraperitoneally injected with
4,5,6,7-tetrabromo-2-(1H-imidazol-2-yl)isoindoline-1,3-dione (tBID)
(10 ng/g; cat. no. HY-100464; MedChemExpress) diluted in 400 µl
PBS. For tBID and tunicamycin (TM; cat. no. HY-A0098;
MedChemExpress) co-treatment, tBID (10 ng/g) and TM (0.5 mg/kg)
were injected intraperitoneally 30 min prior to LPS challenge.
Following this, mice were intraperitoneally injected with 10 mg/kg
LPS (Escherichia coli O111: B4; Sigma-Aldrich; Merck KGaA)
diluted in 300 µl PBS. At 6 h after the LPS injection, mice were
anesthetized with 4% sevoflurane (Abbott Pharmaceutical Co., Ltd.)
and serum (stored in -80˚C) was harvested through the postocular
venous plexus. Cervical dislocation was used for euthanasia.
Preparation of bone marrow-derived
macrophages (BMDMs) and peritoneal macrophages (PMs)
For BMDM generation, mouse femoral tissues were
isolated and the bone marrow was flushed with 3 ml normal saline
(NS). Following red blood cell lysis, bone marrow cells were
resuspended at 2.4x106 cells/ml in DMEM (Hyclone; GE
Healthcare Life Science) supplemented with 10% FBS (Gibco; Thermo
Fisher Scientific, Inc.) and 30 ng/ml granulocyte-macrophage
colony-stimulating factor (PeproTech, Inc.). The culture medium was
changed every 2 days, and after 5-6 days of culturing (37˚C and 5%
CO2), cells were subjected to further experiments.
For PM generation, each mouse was injected
intraperitoneally with thioglycollate (BD Biosciences). At day 3
post-injection, peritoneal lavage was performed with 5 ml NS. Cells
were resuspended at 2-4x106 cells/ml and cultured in
RPMI 1640 (Hyclone; GE Healthcare Life Science) culture medium
supplemented with 10% FBS. LPS stimulation was achieved by adding
LPS (100 ng/ml) at the indicated time points in each experiment,
and the supernatant or mRNA and protein were harvested for further
analysis. TM (10 µg/ml in vitro or 0.5 mg/kg in vivo)
was added 30 min prior to LPS stimulation in vitro and in
vivo.
Reverse transcription-quantitative PCR
analysis
For mRNA analysis, cells were stimulated with LPS
(100 ng/ml) for 0.5, 1, 3, 6 h and then the cells were washed with
PBS and the total RNA were extracted. Total RNA from macrophages
(BMDMs for expression and inhibitor experiments and PMs for siRNA
interference experiments) was extracted using TRIzol®
reagent (cat. no. 10296010; Thermo Fisher Scientific, Inc.).
Complementary DNA templates were obtained by reverse transcription
(50˚C for 45 min and at 85˚C for 5 min) in a 10 µl reaction volume
containing 1 µg total RNA, oligo (dT) primers and a reverse
transcription premix (Takara, Bio, Inc.). Quantitative PCR was
performed using a SYBR-Green PCR system and an ABI 7500 Thermal
Cycler (High-Capacity cDNA Reverse Transcription kit, cat. no.
4374967; PowerUp™ SYBR™ Green Master Mix, cat. no. A25742; Thermo
Fisher Scientific, Inc.) according to the manufacturer's protocol.
SYBR-Green reagents were purchased from Thermo Fisher Scientific,
Inc. The thermocycling conditions were as follows: 95˚C for 3 min,
followed by 40 cycles of denaturation at 95˚C for 10 sec, annealing
at 60˚C for 5 sec and extension at 72˚C for 10 sec. mRNA levels
were normalized to the mRNA level of β-actin, which was used as the
internal control. The primers used were as follows: β-actin,
forward, 5'-CTCCATCCTGGCCTCGCTGT-3' and reverse,
5'-GCTGTCACCTTCACCGTTCC-3'; PERK forward,
5'-TCCCCTAGATCCCCTGAACTT-3' and reverse,
5'-TGGAGTGTCTGATCTTCACTGA-3'; IL-6 forward,
5'-GGCGGATCGGATGTTGTGAT-3' and reverse, 5'-GGACCCCAGACAATCGGTTG-3';
TNF-α forward, 5'-GGAACACGTCGTGGGATAATG-3' and reverse,
5'-GGCAGACTTTGGATGCTTCTT-3'; IRE1α forward,
5'-TTGAGAGAGCTTTTACCAGCAG-3' and reverse,
5'-ACCAGGACCTGACGGATGT-3'; and ATF6, forward,
5'-TGGAGCAGGATGTCCCGTT-3' and reverse,
5'-CTGTGGAAAGATGTGAGGACTC-3'. Relative mRNA levels were determined
using the 2-ΔΔCq method (13) and β-actin was used as the internal
control.
ELISA analysis
Blood was centrifuged at 845 x g for 15 min and
serum was collected. Serum levels of IL-6 and TNF-α were analyzed
using Quantikine IL-6 and TNF-α ELISA kits (cat. nos. M6000B and
MTA00B; R&D Systems, Inc.) according to the manufacturer's
protocol.
Small interfering RNA (siRNA)
interference
Mouse PMs at a confluence of 80% were cultured in
half of the final total culture volume in FBS-free RPMI-1640 or
DMEM and transfected with 3 ng/ml HIPK2 siRNA (Gene Pharma; sense,
5'-GGAGUUCAUUGACCUGUUAAA-3' and anti-sense,
5'-UAACAGGUCAAUGAACUCCCG-3') or 6 ng (3 ng/ml) control siRNA
(Shanghai GenePharma Co., Ltd.; sense, 5'-UUCUCCGAACGUGUCACGUTT-3'
and anti-sense, 5'-ACGUGACACGUUCGGAGAATT-3') in 6-well plates using
INTERFEREin (Invitrogen; Thermo Fisher Scientific, Inc.) for 6 h
(37˚C) according to the manufacturer's protocol. Subsequently, the
other half of the complete culture medium was added and the cells
were cultured (37˚C) for a total of 48 h. After 48 h, cells were
subjected to further stimulation or experiments.
Western blotting
Cells were lysed in RIPA (Beyotime Institute of
Biotechnology). The protein concentration was determined using a
BCA protein assay kit (Thermo Fisher Scientific, Inc.). A total of
30 µg protein/lane was subjected to 10% SDS-PAGE and transferred to
PVDF membranes (Merck KGaA). The membranes were blocked with 5%
non-fat milk in PBS with 0.05% Tween-20 (pH 7.5) at room
temperature for 30 min. Membranes were then immunoblotted with the
primary antibodies (1:2,000) for 4 h at room temperature or at 4˚C
overnight. The primary antibodies used in the present study were:
Anti-mouse HIPK2 (cat. no. 5091; Cell Signaling Technology, Inc.),
phosphorylated (p-) p65 (cat. no. 3033; Cell Signaling Technology,
Inc.), p65 cat. no. 9460; Cell Signaling Technology, Inc.),
phosphorylated (p-)PERK (cat. no. 3179S; Cell Signaling Technology,
Inc.), PERK (cat. no. 3192S; Cell Signaling Technology, Inc.),
p-IRE1α (cat. no. ab48187; Abcam), IRE1α (cat no. 3294S; Cell
Signaling Technology, Inc.), ATF6 (cat. no. ab122897; Abcam) and
β-actin (cat. no. ab8227; Abcam). Subsequently, membranes were
incubated with horseradish peroxidase-conjugated secondary
antibodies (1:3,000) (cat. nos. 7074 and 7076; Cell Signaling
Technology, Inc.). Finally, the blots were visualized using
SuperSignal™ West Atto Ultimate Sensitivity Substrate (Thermo
Fisher Scientific, Inc.).
Statistical analysis
Data from three independent experiments were
repeated and are presented as the mean ± standard deviation.
Student's t-test was used to analyze differences between two
groups. One-way ANOVA was used to analyze differences among
multiple groups. Sidak's multiple comparisons test or Dunnett's
post hoc test were used as the post hoc tests. Analysis was
performed with Prism software (version no. 8; GraphPad Software,
Inc.). P<0.05 was considered to indicate a statistically
significant difference.
Results
HIPK2 expression is upregulated by LPS
stimulation in macrophages
To investigate the expression levels of HIPK2 during
inflammatory cytokine production in macrophages, the mRNA and
protein expression levels of cytokines and HIPK2 were analyzed. The
results demonstrated that following LPS stimulation, IL-6 and TNF-α
were significantly upregulated at the mRNA (Fig. 1A and B) and protein (Fig. 1C and D) levels. The peak transcription of IL-6
and TNF-α was observed at 3 h following stimulation. Furthermore,
the expression levels of HIPK2 were elevated following LPS
stimulation (Fig. 1E and F), and the peak production was observed at
0.5 and 1 h after LPS stimulation at the mRNA and protein levels,
respectively. These results demonstrated that HIPK2 expression is
upregulated earlier than the expression of cytokines, indicating a
potential role of HIPK2 in the regulation of cytokine production in
macrophages.
HIPK2 knockdown suppresses IL-6 and
TNF-α production in macrophages
The potential role of HIPK2 in the cytokine
production by macrophages was investigated. siRNA was used to knock
down HIPK2 expression, and the effects on macrophages were
investigated. The results revealed that HIPK2 was successfully
knocked down at the mRNA level in LPS stimulation (Fig. 2A) and protein (Fig. 2B) levels. Furthermore, the results
demonstrated that the phosphorylation of p65, the effective
component of NF-κB (3), was
markedly downregulated following HIPK2 knockdown (Fig. 2B) in the presence of LPS. Similarly,
IL-6 (Fig. 2C) and TNF-α (Fig. 2D) production was suppressed
following HIPK2 knockdown. Therefore, these results indicated that
HIPK2 may serve an essential role in cytokine production during LPS
stimulation in macrophages.
HIPK2 knockdown suppresses ER stress
in LPS-stimulated macrophages
The mechanism by which HIPK2 promoted cytokine
production was investigated. A recent study has demonstrated the
crucial role of ER stress in macrophage inflammation (6). Inhibition of ER stress alleviates
inflammation and cytokine production by macrophages (7). The results demonstrated that, after
the interference of HIPK2, the mRNA level of IRE1α in 1 and 3 h
after LPS stimulation, PERK and ATF6 in 3 h after LPS stimulation
were downregulated (Fig. 3A-C). The
results also revealed that the protein level of p-IRE1α, p-PERK and
ATF6 at 15, 30 and 60 min after the LPS stimulation were
downregulated (Fig. 3D), indicating
that ER stress may be a potent mechanism of HIPK2 for the promotion
of cytokine production by macrophages.
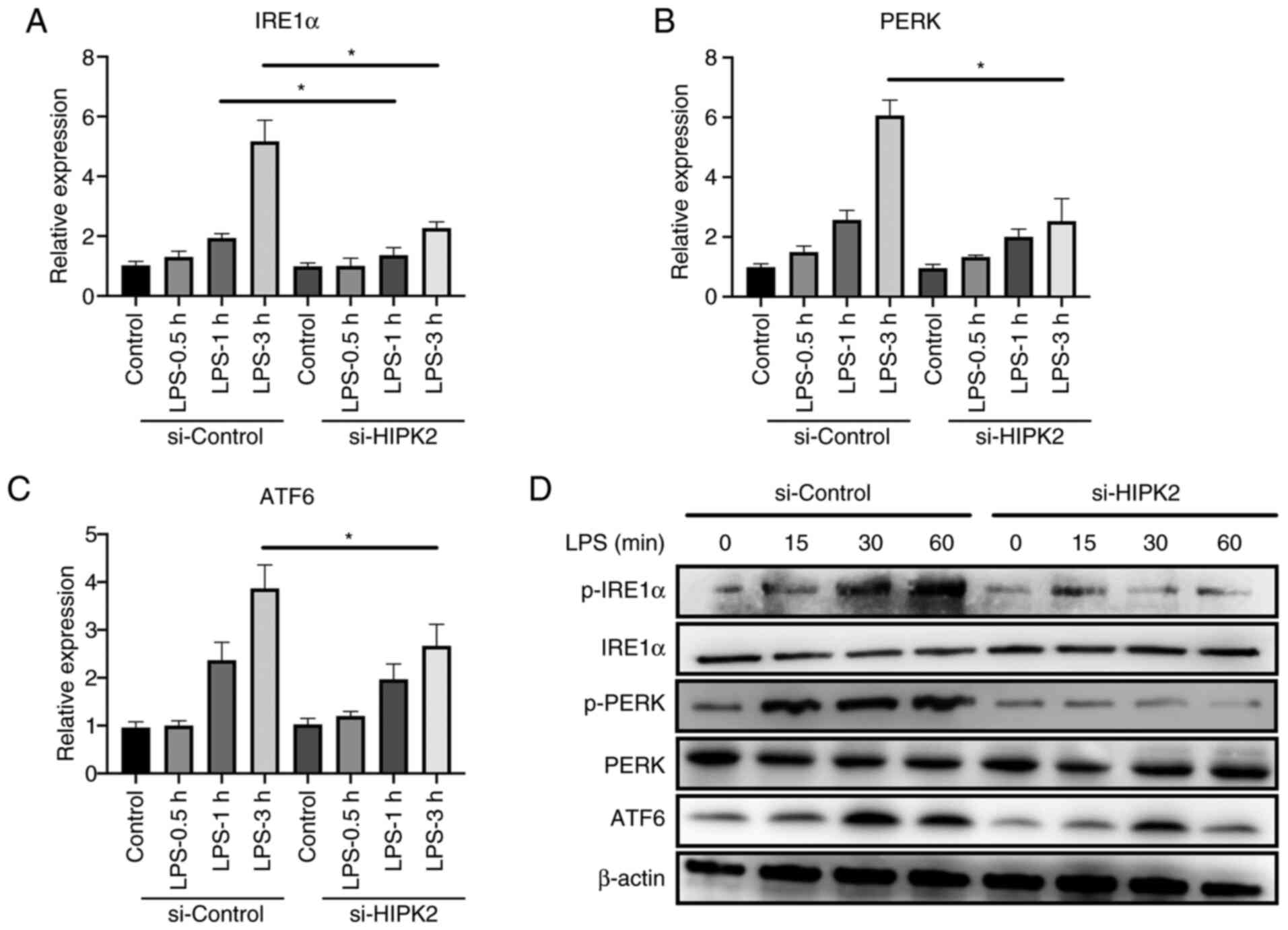 | Figure 3HIPK2 knockdown suppresses endoplasmic
reticulum stress in LPS-stimulated macrophages. Reverse
transcription-quantitative PCR analysis of (A) IRE1α, (B) PERK and
(C) ATF6 expression following HIPK2 interference in LPS-stimulated
PMs (n=3). (D) Western blot analysis of p-IRE1α, IRE1α, p-PERK,
PERK and ATF6 following HIPK2 interference in LPS-stimulated PMs.
*P<0.05 as indicated. HIPK2, homeodomain-interacting
protein kinase 2; LPS, lipopolysaccharides; IRE1α,
inositol-requiring enzyme 1α; PERK, PKR-like endoplasmic reticulum
kinase; ATF6, activating transcription factor 6; PMs, peritoneal
macrophages; p-, phosphorylated; si, small interfering RNA. |
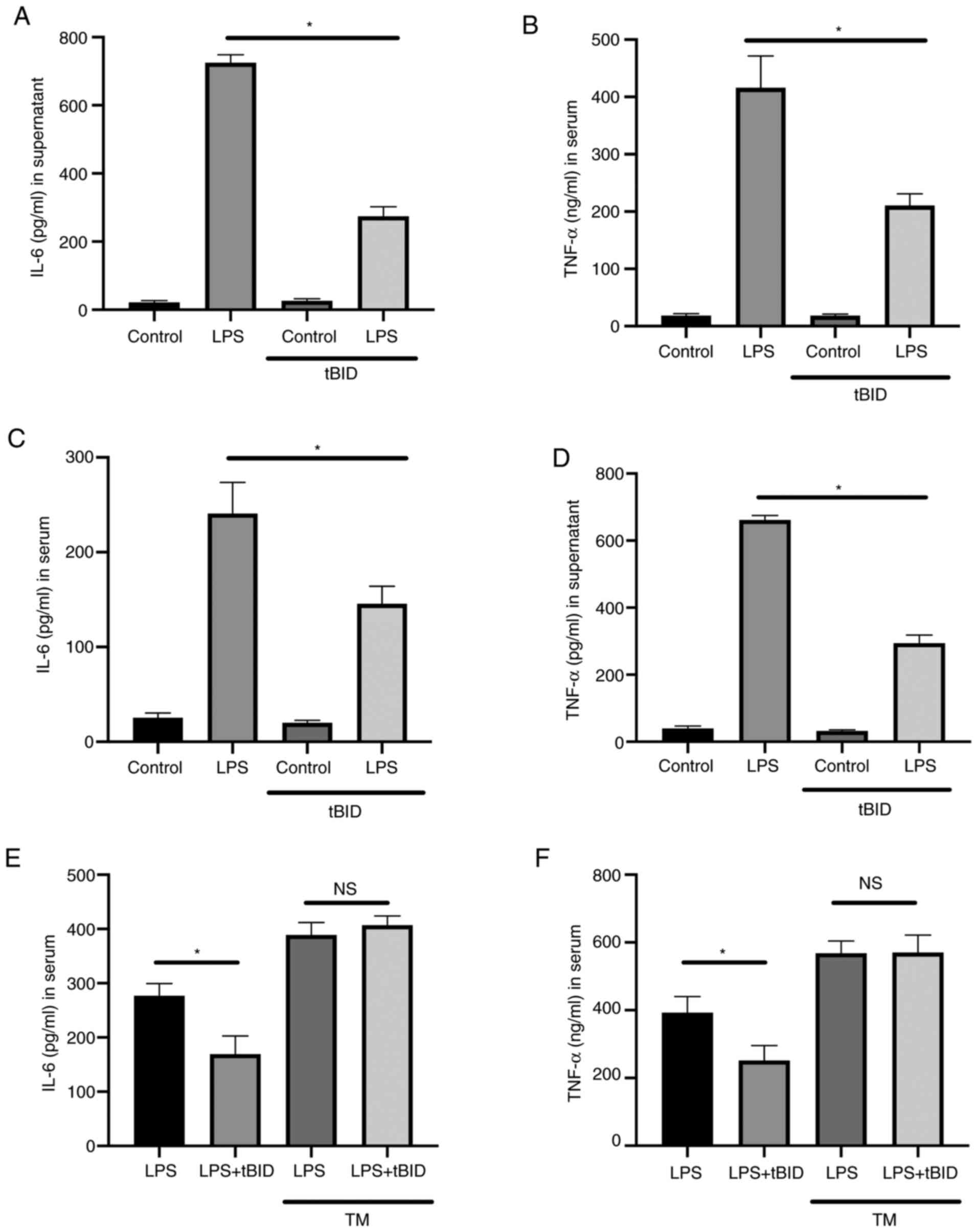
Activation of ER stress restores IL-6
and TNF-α production following HIPK2 knockdown in LPS-stimulated
macrophages
To investigate the essential role of ER stress in
HIPK2-mediated cytokine production, TM, an ER stress agonist
(14), was used to establish the
effects of HIPK2 knockdown in macrophages. The results revealed
that while HIPK2 knockdown attenuated IL-6 and TNF-α production,
the application of TM, though slightly decreased IL-6 and TNF-α
compared with the control, abolished the attenuation of cytokine
production (Fig. 4A and B) and restored the phosphorylation level
of p65 (Fig. 4C). Therefore, these
results indicated that ER stress is an essential process for
HIPK2-mediated cytokine production by macrophages.
HIPK2 inhibition suppresses IL-6 and
TNF-α production in vitro and in vivo
To investigate whether HIPK2 was a potent
anti-inflammatory agent. The effects of the HIPK2 inhibitor, tBID,
in LPS-stimulated macrophages and LPS-challenged mice were
investigated. The results demonstrated that, in cultured
macrophages, tBID treatment suppressed IL-6 (Fig. 5A) and TNF-α (Fig. 5B) levels in the LPS treatment
groups. Furthermore, in LPS-challenged mice, inhibition of HIPK2
was associated with favorable outcomes in terms of attenuating the
serum levels of IL-6 (Fig. 5C) and
TNF-α (Fig. 5D). TM was used to
activate ER stress in the presence of tBID to investigate whether
the ER agonist restored cytokine production following HIPK2
inhibition. The results demonstrated that IL-6 (Fig. 5E) and TNF-α (Fig. 5F) levels were elevated compared with
the LPS group without TM treatment, and the effects of HIPK2
inhibition were decreased by TM treatment. The TM treatment was
also demonstrated to elevate IL-6 and TNF-α levels (Fig. 5E and F). Collectively, these results indicated
that HIPK2 inhibition suppressed cytokine production in
vitro and in vivo, suggesting its potential role in
anti-inflammation management.
Discussion
Uncontrolled activation of inflammation and
subsequent cytokine storm are essential contributors to
inflammatory organ dysfunction and failure (15). Endotoxemia and sepsis are usually
initiated from the overactivation of macrophages and the following
excessive production of inflammatory cytokines (4,16).
Therefore, an in-depth understanding of cytokine production
regulation may provide an important mechanism for the management of
inflammation. The present study revealed that HIPK2 served a
positive role in maintaining LPS-induced inflammation of
macrophages and that HIPK2 knockdown led to the attenuation of
cytokine production. Two types of macrophages were used in the
present study. BMDMs and PMs are regarded as models for macrophage
investigation (2,3). The present study used BMDMs to analyze
the expression levels of HIPK2 in in vitro inhibitor
experiments and in in vitro experiments with HIPK2
interference, while PMs were used to ensure the interference
efficacy (4). The results were in
accordance with a previous study that reported that HIPK2 was
crucial in maintaining type I interferon production in antiviral
immunity of macrophages (12). The
present study highlighted that HIPK2 may be a potential target to
control excessive cytokine production by overactivated
macrophages.
The results demonstrated that ER stress served an
essential role in HIPK2-mediated cytokine production. Through HIPK2
knockdown and ER stress agitation, the present study revealed that
HIPK2-promoted cytokine production was dependent on the activation
of ER stress, indicating that HIPK2 may activate ER stress to
promote cytokine production. ER stress has been reported to serve a
crucial role in macrophagic inflammation (17). Previous studies have demonstrated
that the activation of ER stress promotes NF-κB and Nrf2
activation, which further promotes the transcription of
pro-inflammatory genes, such as p65 and Erk, and oxidative stress
(6,9). Additionally, ER stress promotes
mitochondrial stress and initiates respiratory burst in
mitochondria, promoting the production of reactive oxygen species
in macrophages (18-20).
Numerous molecules, such as ALDH2 and NOD1, regulate ER stress
through exogenous receptor-ligand reactions and intrinsic
regulation (20,21). The present study indicated a
potential role in regulating ER stress. Investigation of the
specific mechanism of the regulation could be a topic of interest
in future studies. However, for the in vivo experiments,
since the present study was not able to select targeted
macrophages, the alteration of these markers could not be measured.
LPS challenge results in systematic inflammation and macrophages
and monocyte-macrophages can infiltrate organs and tissues
(22). This is in accordance with a
previous study which demonstrated the in vivo expression of
certain macrophage genes, including v-ets erythroblastosis virus
E26 oncogene homolog 1 and 2, and presented the results as serum
cytokine levels following the inhibition or activation of various
molecules, including Erk or interleukin 1 receptor-associated
kinase M (3), which the present
study investigated using tBID and TM in vivo.
Since cytokine production relies on HIPK2 expression
in macrophages, the present study investigated the potential
effects of HIPK2 inhibition in an LPS-stimulated model in
vitro and in vivo. However, although the present study
was a preliminary investigation and further studies are required to
address the effects in different inflammatory models, the present
study indicated the therapeutic effects of HIPK2 inhibition in
anti-inflammation management. HIPK2 may be a promising target for
the treatment of inflammatory diseases.
In summary, the present study reported the crucial
role of HIPK2 in promoting cytokine production in response to LPS
in macrophages. Furthermore, the results demonstrated that
HIPK2-mediated cytokine production was dependent on the activation
of ER stress. Targeting HIPK2 may be a potential strategy for the
management of inflammation.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant nos. 8167080319 and
81701899).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
LX and HF performed the majority of experiments and
prepared the manuscript. DX assisted in animal experiments and
contributed to the preparation of the manuscript. GW designed the
present study and reviewed the manuscript. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
The Ethics Committee of Changhai Hospital approved
the animal experiments.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Thorburn AN, Macia L and Mackay CR: Diet,
metabolites, and ‘western-lifestyle’ inflammatory diseases.
Immunity. 40:833–42. 2014.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Lamkanfi M, Sarkar A, Vande Walle L,
Vitari AC, Amer AO, Wewers MD, Tracey KJ, Kanneganti TD and Dixit
VM: Inflammasome-dependent release of the alarmin HMGB1 in
endotoxemia. J Immunol. 185:4385–4392. 2010.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Ma X, Jiang Z, Li N, Jiang W, Gao P, Yang
M, Yu X, Wang G and Zhang Y: Ets2 suppresses inflammatory cytokines
through MAPK/NF-κB signaling and directly binds to the IL-6
promoter in macrophages. Aging (Albany NY). 11:10610–10625.
2019.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Xu M, Jiang Z, Wang C, Li N, Bo L, Zha Y,
Bian J, Zhang Y and Deng X: Acetate attenuates inflammasome
activation through GPR43-mediated Ca(2+)-dependent NLRP3
ubiquitination. Exp Mol Med. 51(83)2019.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Wen M, Ma X, Cheng H, Jiang W, Xu X, Zhang
Y, Zhang Y, Guo Z, Yu Y, Xu H, et al: Stk38 protein kinase
preferentially inhibits TLR9-activated inflammatory responses by
promoting MEKK2 ubiquitination in macrophages. Nat Commun.
6(7167)2015.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Zeeshan HM, Lee GH, Kim HR and Chae HJ:
Endoplasmic reticulum stress and associated ROS. Int J Mol Sci.
17(327)2016.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Sicari D, Delaunay-Moisan A, Combettes L,
Chevet E and Igbaria A: A guide to assessing endoplasmic reticulum
homeostasis and stress in mammalian systems. FEBS J. 287:27–42.
2020.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Fu S, Yang L, Li P, Hofmann O, Dicker L,
Hide W, Lin X, Watkins SM, Ivanov AR and Hotamisligil GS: Aberrant
lipid metabolism disrupts calcium homeostasis causing liver
endoplasmic reticulum stress in obesity. Nature. 473:528–531.
2011.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Lebeaupin C, Vallée D, Hazari Y, Hetz C,
Chevet E and Bailly-Maitre B: Endoplasmic reticulum stress
signalling and the pathogenesis of non-alcoholic fatty liver
disease. J Hepatol. 69:927–947. 2018.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Akaike Y, Kuwano Y, Nishida K, Kurokawa K,
Kajita K, Kano S, Masuda K and Rokutan K: Homeodomain-interacting
protein kinase 2 regulates DNA damage response through interacting
with heterochromatin protein 1γ. Oncogene. 34:3463–3473.
2015.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Torrente L, Sanchez C, Moreno R, Chowdhry
S, Cabello P, Isono K, Koseki H, Honda T, Hayes JD, Dinkova-Kostova
AT and de la Vega L: Crosstalk between NRF2 and HIPK2 shapes
cytoprotective responses. Oncogene. 36:6204–6212. 2017.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Cao L, Yang G, Gao S, Jing C, Montgomery
RR, Yin Y, Wang P, Fikrig E and You F: HIPK2 is necessary for type
I interferon-mediated antiviral immunity. Sci Signal.
12(eaau4604)2019.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Xu Z, Duan F, Lu H, Abdulkadhim Dragh M,
Xia Y, Liang H and Hong L: UBIAD1 suppresses the proliferation of
bladder carcinoma cells by regulating H-Ras intracellular
trafficking via interaction with the C-terminal domain of H-Ras.
Cell Death Dis. 9(1170)2018.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Zhang XW, Meng X, Chen YY, Leng SX and
Zhang HY: The biology of aging and cancer frailty, inflammation,
and immunity. Cancer J. 23:201–205. 2017.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Jiang Z, Bo L, Meng Y, Wang C, Chen T,
Wang C, Yu X and Deng X: Overexpression of homeodomain-interacting
protein kinase 2 (HIPK2) attenuates sepsis-mediated liver injury by
restoring autophagy. Cell Death Dis. 9(847)2018.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Kim O-K, Jun W and Lee J: Mechanism of ER
stress and inflammation for hepatic insulin resistance in obesity.
Ann Nutr Metab. 67:218–227. 2015.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Levada K, Guldiken N, Zhang X, Vella G, Mo
FR, James LP, Haybaeck J, Kessler SM, Kiemer AK, Ott T, et al:
Hsp72 protects against liver injury via attenuation of
hepatocellular death, oxidative stress, and JNK signaling. J
Hepatol. 68:996–1005. 2018.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Li L, Wang H, Zhang J, Sha Y, Wu F, Wen S,
He L, Sheng L, You Q, Shi M, et al: SPHK1 deficiency protects mice
from acetaminophen-induced ER stress and mitochondrial permeability
transition. Cell Death Differ. 27:1924–1937. 2020.PubMed/NCBI
|
|
20
|
Keestra-Gounder AM, Byndloss MX, Seyffert
N, Young BM, Chávez-Arroyo A, Tsai AY, Cevallos SA, Winter MG, Pham
OH, Tiffany CR, et al: NOD1 and NOD2 signalling links ER stress
with inflammation. Nature. 532:394–397. 2016.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Kimura Y, Inoue A, Hangai S, Saijo S,
Negishi H, Nishio J, Yamasaki S, Iwakura Y, Yanai H and Taniguchi
T: The innate immune receptor Dectin-2 mediates the phagocytosis of
cancer cells by Kupffer cells for the suppression of liver
metastasis. Proc Natl Acad Sci USA. 113:14097–14102.
2016.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Pang J, Peng H, Wang S, Xu X, Xu F, Wang
Q, Chen Y, Barton LA, Chen Y, Zhang Y and Ren J: Mitochondrial
ALDH2 protects against lipopolysaccharide-induced myocardial
contractile dysfunction by suppression of ER stress and autophagy.
Biochim Biophys Acta Mol Basis Dis. 1865:1627–1641. 2019.PubMed/NCBI View Article : Google Scholar
|