Introduction
Limb-girdle muscular dystrophies (LGMDs) are a group
of autosomal hereditary diseases characterized by progressive
muscle weakness in the scapular and pelvic girdle and trunk
muscles. LGMDs were firstly classified into autosomal dominant and
autosomal-recessive types by the European Neuromuscular Center in
1995(1). The autosomal-recessive
form of LGMD type 2A [LGMD2A; Online Mendelian Inheritance in Man
(OMIM) ID, 253600), which is also referred to as limb girdle
muscular dystrophy type R1 (LGMDR1) and caused by variant in the
calpain-3 (CAPN3) gene (OMIM ID, 114240), is the most common type
of LGMDs, accounting for ~30% of all cases of LGMD (2). However, a 21-bp deletion in the CAPN3
gene was also identified to cause the autosomal-dominant type of
LGMD2A (3). According to the
updated 2018 classification for LGMD, there are 5
autosomal-dominant and 24 autosomal-recessive subtypes, most of
which are due to genetic defects associated with severe congenital
muscular dystrophy (4). The
incidence of LGMD2A is ~1 in 100,000 individuals and the average
age of onset is 17.9 years (5). The
clinical characteristics of LGMD2A are highly heterogeneous and not
only is there variation in the age of onset (from early childhood
to adulthood) and the degree of muscular weakness, but also in the
high prevalence of variability among gene variants. Clinical
features include difficulties in walking and running, a waddling
gait, scapular winging and respiratory failure in the advanced
stages of the disease; however, the facial and neck muscles are not
affected (6). In addition, cases
involving cardiac muscles have been reported at a low frequency
(7,8).
The CAPN3 gene consists of 24 exons and encodes the
CAPN3 enzyme. CAPN3, a Ca2+-dependent protease, is
composed of 821 amino acids and is involved in the breakdown and
cleavage of a variety of key skeletal myoproteins, particularly
those related to the skeletal structure of myofibrils (9). CAPN3 contains four domains: The
protease core subdomain 1, protease core subdomain 2, C2
domain-like domain and penta-EF-hand domain (10). Upon Ca2+ binding to the
protease core subdomains 1 and 2, they fold into a structurally
active ‘CysPc’ domain, which is a CAPN3-like cysteine protease
sequence motif as defined in the conserved domain database of the
National Center for Biotechnology Information (11). CAPN3 has an important role in LGMD
due to the physiological aberrations caused by its defect, such as
muscular weakness, as a result of the lack of ryanodine receptor
regulation by CAPN3(9).
The present study reported on two Chinese pedigrees,
including six patients affected with LGMD2A caused by CAPN3 gene
variants. Whole-exome sequencing (WES) was performed to make an
accurate diagnosis, which provided a basis for the genetic
counseling of the two families. Furthermore, by analyzing the
genetic patterns of these two families, the pathogenic
classification of the variants in the present study was further
clarified to enhance the current knowledge on the pathology,
facilitating future clinical management.
Materials and methods
Patients and DNA extraction
From family 1 (F1), three patients, their mother and
three healthy siblings were recruited in Tianjin Children's
Hospital (Tianjin, China) in September 2018. From F2, 15 family
members recruited in Tianjin Children's Hospital were studied in
total in June 2019. Genomic DNA was extracted from the peripheral
blood using a Blood Genomic DNA Mini kit (cat. no. CW0541; CoWin
Biosciences) according to the manufacturer's protocol. The ratio of
the absorbance at 260 and 280 nm (A260/280 ratio) were evaluated
with 1 µl of DNA extraction using the NanoDrop® 2000
spectrophotometer (Thermo Fisher Scientific, Inc.). A total of 100
µl DNA solution (≥10 ng/µl) was obtained, which was stored at
-20˚C. Written informed consent was obtained from all family
members and the study was approved by the ethics committee of
Tianjin Children's Hospital (Tianjin, China).
WES and bioinformatics analysis
WES for patient 1 (F1-II11) and patient 4 (F2-II11)
was performed by BGI Group. Paired-end sequencing was performed
with read lengths of 150 bp and an average coverage depth of
100-fold in >95% of the target regions, including all coding
regions and exon-intron boundaries. Sequencing data were aligned to
the human reference genome hg19 using the Burrows-Wheeler Aligner
software. Genome Analysis Toolkit software was used to analyze the
insertion, deletion and single nucleotide polymorphism sites.
Variant annotations were made using the ANNOVAR tool (V20180118;
https://doc-openbio.readthedocs.io/projects/annovar/en/latest/),
1,000 genomes (https://www.1000genomes.org/), dbSNP (https://www.ncbi.nlm.nih.gov/snp/?term=)
and OMIM (https://omim.org/) databases. The effect
of the variants on the structure and function of the proteins was
predicted using Polymorphism Phenotyping v2 software (http://genetics.bwh.harvard.edu/pph2/index.shtml)
and Sorts Intolerant From Tolerant software (V1.1; http://sift.jcvi.org,). In addition, Human Splicing
Finder (V3.1; http://www.ummd.be/HSF/) was used to
predict the splice sites in the gene.
Variant screening and Sanger
sequencing
PCR and further Sanger sequencing were performed on
all patients and other available family members to confirm the
candidate variants and analyze the co-segregation pattern. The
relative exons and flanking intron regions were amplified using
primers designed by Oligo7(12)
(version 7.60; exon 3, 5'-CCCCAAACACAAAATAGGATG-3' and
3'-CACATATGCACGTATAGAGG-5'; exon 10, 5'-GCCACCCTCTTTTCATCCTCC-3'
and 3'-TGTTCCCACAGTTTCCTGCTTC-5'; exon 11,
5'-TGTAGGGAATAGAAATAAATGG-3' and 3'-CCAGGAGCTCTGTGGGTCA-5'; and
another pair of primers for exon 11, 5'-AGAATGAAAGCCCAGAGAGGA-3'
and 3'-TGTGGGTCACTGGGTATTGA-5'). Amplification was performed in a
final volume of 50 µl, containing 25 µl 2X GC buffer I, 20 mM
deoxynucleoside triphosphates mixture, 100-200 ng DNA, 0.5 µM
forward and reverse primers and 2.5 U LA Taq polymerase (cat. no.
RR02AG; Takara Biotechnology Co., Ltd.). The following
thermocycling conditions were used for the PCR: Initial
denaturation for 2 min at 94˚C, followed by 35 cycles of 94˚C for
30 sec, 58˚C for 30 sec and 72˚C for 40 sec; and a final extension
step at 72˚C for 5 min. The PCR products were separated by 1.5%
agarose gel electrophoresis and the proper DNA was purified from
agarose gel using a Gel Extraction kit (CoWin Biosciences) and sent
to Genewiz (Beijing, China) for Sanger sequencing. Chromas software
(version 1.62; Technelysium Pty Ltd.) was used to compare the
sequencing data with the reference sequences (NM_000070.2) in
GenBank (https://www.ncbi.nlm.nih.gov/nuccore/NC_000015.10?report=genbank&from=42359501&to=42412317)
to determine the variants.
Results
Patients
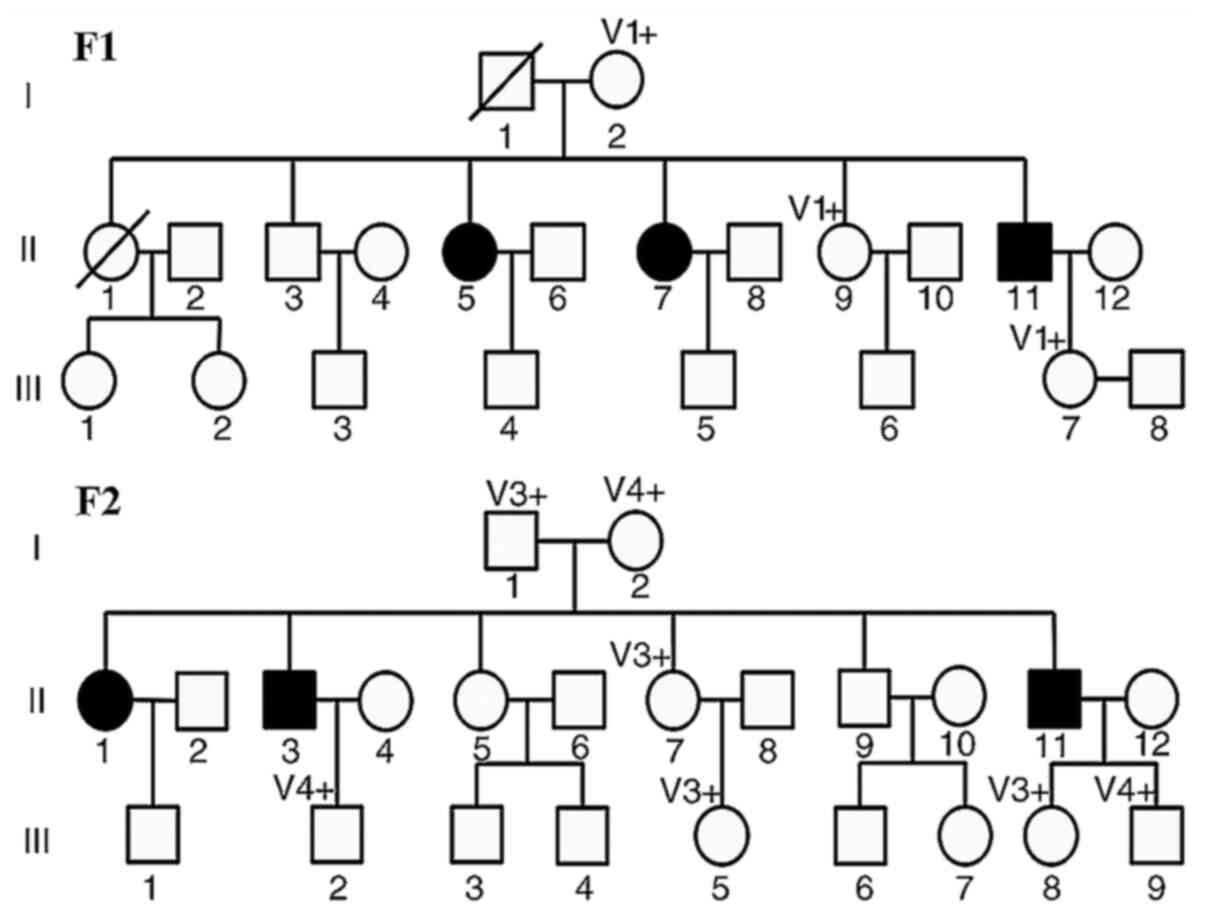
F1 comprised three affected family members born from
non-consanguineous Chinese parents (Fig. 1). Patient 1 (F1-II11) was a
58-year-old male who experienced difficulties running and jumping
from the age of 9 years. At the age of 19 years, F1-II11 frequently
fell down and required help to walk upstairs. Gradually, the
patient was no longer able to raise his arms and relied on a
wheelchair from the age of 46 years. Approximately 30 years
previously, the patient underwent general laboratory tests and
imaging examinations; the serum creatine kinase (CK) levels were
560 U/l (normal, <308 U/l) and needle electromyography revealed
fibrillation potentials in the scapular girdle muscles. However,
the electrocardiogram was normal. Subsequently, F1-II11 was
misdiagnosed with Becker muscular dystrophy (BMD). Patient 2
(F1-II7) is a 60-year-old woman, the elder sister of patient 1 who
is unable to walk since she was 20 years old and currently relies
on a wheelchair for mobility. Patient 3 (F1-II5) is 62 years old,
the elder sister of patients 1 and 2 who uses a wheelchair but is
able to walk on crutches. The only daughter (F1-Ⅲ7) of patient 1,
who is healthy, wished to have a healthy child, and therefore
attended Tianjin Children's Hospital (Tianjin, China) for genetic
counseling and pedigree gene screening.
In F2, there were three affected patients (Fig. 1). Patient 4 (F2-II11) was a
37-year-old male; the age of disease onset was 30 years, when the
patient first experienced limb muscle weakness and difficulties
running, jumping and walking up stairs. At the age of 35 years,
F2-II11 was unable to walk independently and a laboratory
examination revealed high levels of serum CK (1,609 U/l). The
patient's symptoms were progressively aggravating; he presented
with winged scapulae and is dependent on a wheelchair (Fig. 2A and B). Patient 5 (F2-II3) is 45 years old, the
elder brother of patient 4, who first experienced muscle weakness
at the age of 26 years. After three years, F2-II3 was unable to
walk, stand up, stoop or hold objects with his hands. F2-II3 also
presented with winged scapulae (Fig.
2C and D). Electromyography
also revealed that the muscle tension was low and the serum CK
levels were elevated to 1,544 U/l. Patient 6 (F2-II1), a
50-year-old female, is the elder sister of patients 4 and 5.
Patient 6 was 20 years old when symptoms of limb weakness first
appeared. However, she was misdiagnosed with rheumatoid arthritis.
After 20 years, patient 6 was unable to walk and became dependent
on a wheelchair. The wife of patient 4 wished to know about her
children's health, so she opted for genetic counseling and pedigree
gene screening.
CAPN3 gene variants. F1
The spectrum of clinicopathological and genetic
features is presented in Table I.
F1 comprised a three-generation pedigree containing three patients
and four unaffected controls. The WES results revealed that the
compound heterozygous variants of the CAPN3 gene (NM_000070.2) were
responsible for LGMD2A in F1. The heterozygous variant of an A-to-G
transition located in -9 upstream of exon 10 of CAPN3 (c.1194-9
A>G) was identified, which has been reported previously and
classified as ‘pathogenic’ in the ClinVar database. Another
heterozygous variant was identified in exon 11 [c.1437C>T
(p.Ser479=)], which caused the 479th genetic codon change from AGC
to AGT; however, the amino acid serine did not change. To the best
of our knowledge, this synonymous variant has not been reported in
the previous literature. According to the 2015 American College of
Medical Genetics and Genomics (ACMG) variant classification
guidance (13), the c.1437C>T
(p.Ser479=) variation may be classified as ‘likely pathogenic’.
 | Table IClinical and genetic characteristics
of patients. |
Table I
Clinical and genetic characteristics
of patients.
| Patient | Sex | AOO (years) | DOWD (years) | SW | CPH | Gene | CK level (U/l) |
|---|
|
F1-II11 | M | 9 | 37 | No | NA | CAPN3 | 560 |
|
F1-II7 | F | 12 | 15 | No | NA | CAPN3 | NA |
|
F1-II5 | F | 20 | 35 | No | NA | CAPN3 | NA |
|
F2-II11 | M | 30 | 5 | Yes | NA | CAPN3 | 1609 |
|
F2-II3 | M | 26 | a | Yes | NA | CAPN3 | 1544 |
|
F2-II1 | F | 20 | 20 | No | NA | CAPN3 | NA |
F2
The WES results revealed the presence of compound
heterozygous variants of the CAPN3 gene in F2. The c.1468C>T
(p.Arg490Trp) variant in exon 11 was listed in the ClinVar database
and it is regarded as a ‘pathogenic/likely pathogenic’ variant. The
c.632+4A>G variant was identified in the donor site of intron 4,
which is regarded as a variant of ‘uncertain significance’ in the
ClinVar database.
No other variations associated with muscular
dystrophy were observed from the results of the WES analysis in the
two families. Although the two families had no intention to undergo
further muscle biopsy, as they only sought genetic counseling and
pedigree gene screening, the diagnosis of LGMD2A was possible
according to the results of WES (all patients carried the compound
heterozygous variants of the CAPN3 gene) combined with the clinical
manifestations in the patients in the two families.
Sanger sequencing analysis. F1
According to the result of the WES, the DNA of three
patients and another four healthy family members was amplified. The
results of the Sanger sequencing were consistent with those of WES
(Fig. 3). Sanger sequencing
demonstrated that the variant c.1194-9A>G was inherited from the
mother (F1-I2), whereas the c.1437C>T (p.Ser479=) variant was
not present in the maternal sequencing results. The genotype of the
patients' father could not be verified due to his prior death.
Patients 2 and 3 were both in the same compound heterozygous state.
The sister (F1-II9) and daughter (F1-Ⅲ7) of patient 1 carried the
c.1194-9 A>G variant. In the son-in-law of proband 1, the
aforementioned variants were not detected.
F2
The DNA of patients 4, 5 and 6 and another 12 family
members was amplified. The Sanger sequencing results revealed that
the c.632+4A>G variant was inherited from the proband's father
(F2-I1) and the c.1468C>T (p.Arg490Trp) variant was inherited
from the mother (F2-I2). It also identified that patients 4, 5 and
6 were all carriers of the compound heterozygous variants. F2-II7,
F2-Ⅲ5 and F2-Ⅲ8 carried the c.632+4 A>G variant, while F2-Ⅲ2 and
F2-Ⅲ9 carried the c.1468C>T (p.Arg490Trp) variant. The remaining
five family members did not carry either of the variants (Fig. 4).
Discussion
LGMDs are a heterogeneous group of neuromuscular
diseases associated with weakness in the proximal muscles, which
vary in terms of age of onset, clinical severity, genotype,
phenotype and duration of disease prior to requiring wheelchair
assistance. LGMD2A is the most common subtype of LGMD; it may
affect individuals at any age and exhibits variable clinical
features, ranging from mild to severe forms. While it may occur at
any age from 2 to 55 years, the mean age of onset is 17.9 years
(5). In addition, the mean age of
patients relying on wheelchair usage is 35.2 years and the average
duration from disease onset to wheelchair dependence is 15.2 years,
but it ranges from 4 to 29 years (5). In the present study, the minimum age
of onset was 9 years (patient 1) and the maximum was 30 years
(patient 4). In addition, the longest duration from disease onset
to becoming wheelchair-dependent was 37 years in patient 1, while
the shortest was 5 years in patient 4. Thus, these results revealed
a wide variation in disease progression. The mean period was 22.4
years in the present study; at the early stage, muscle weakness may
occur in the pelvic and shoulder girdle. Individuals with LGMD2A
may have an unusual walking gait and may experience difficulty
walking, running and raising the arms. Toe-walking may also be
present in early childhood before muscle weakness is detected
(14). As the disease progresses,
patients may lose the ability to walk and therefore, eventually
become dependent on wheelchair use. In general, LGMD2A
predominantly affects the proximal muscles and not the brain.
However, additional features such as respiratory failure and severe
cardiomyopathy have been reported in specific cases (6-8).
In the first pedigree examined in the present study,
patient 1 was misdiagnosed with BMD previously. According to the
pedigree study, 2 cases appeared in females in generation II, but
none of the males in generation III was affected. BMD is an
X-linked dystrophinopathy, of which the age of onset is usually
late and the disease progress is slow; the majority of the patients
are still able to walk at the age of 30 years (15). Therefore, it is easy to dismiss the
previous diagnosis according to the family inheritance pattern and
the clinical features. WES revealed that the heterozygous variant
of c.1194-9A>G was present in the 9th intron, which was
classified as ‘pathogenic’ in the ClinVar database. This
substitution leads to a new splice site in position 9 upstream of
exon 10, creating a new AG acceptor splice site and the retention
of the eight last nucleotides of intron 9(16). Another heterozygous variant,
c.1437C>T (p.Ser479=), was identified as a synonymous variant in
exon 5, which caused the genetic codon to change, but not the amino
acid from serine. In the 1000 Genomes and gnomAD databases, the
frequency of this synonymous variant in the East Asian population,
which did not belong to a single nucleotide polymorphism site, was
recorded as 3 and 2.5%, respectively, and the frequency in the
whole population was 6 per 10,000 and 1 per 10,000, respectively.
It was classified as a variant of ‘uncertain significance’ in the
ClinVar database. Subsequently, the online software Human Splicing
Finder (http://www.ummd.be/HSF/) was used to
predict gene splice sites. The results revealed that the score of
the variant was 81.49 above the threshold; thus, it may be
considered to be a splice site acceptor. According to the ACMG
variant classification guidance (13), the c.1437C>T (p.Ser479=) variant
was consistent with the pathogenic evidence of PM2 [absent from
controls (or at extremely low frequency if recessive) in the Exome
Sequencing Project, 1000 Genomes Project or Exome Aggregation
Consortium], PM3 (for recessive disorders, detected in trans with a
pathogenic variant), PP1 (co-segregation with disease in multiple
affected family members in a gene definitively known to cause the
disease), PP3 (multiple lines of computational evidence support a
deleterious effect on the gene or gene product) and PP4 (patient's
phenotype or family history is highly specific for a disease with a
single genetic etiology); thus, the synonymous variant was
classified as ‘likely pathogenic’. Previously, it was thought that
synonymous variants had no functional effect; however, it has been
reported that synonymous variants may affect pre-mRNA splicing by
generating new splice sites or interfering with the original site
and affecting the efficiency of translation owing to the alteration
of the mRNA secondary structure (17,18).
In the second pedigree, the heterogeneous p.A490T
variant in exon 11 was determined to be a ‘pathogenic/likely
pathogenic’ variant in the ClinVar database. The other
heterogeneous variant was in the donor site of intron 4, which was
classified as being a variant of ‘uncertain significance’ in the
ClinVar database. This splice site variant was reported in 2008;
Blazquez et al (19)
amplified CAPN3 complementary DNA and discovered that the A to G
change promoted the skipping of exon 4 in the mRNA. According to
the ACMG variant classification criteria, the c.632+4A>G variant
may be classified as ‘likely pathogenic’. Based on the WES analysis
along with Sanger sequencing, which confirmed the co-segregation
among the family members, it may be concluded that the compound
heterogeneous variants were the cause of LGMD2A in the two
pedigrees. In fact, there are significant differences in ACMG
classifications determined by different laboratories and
clinicians; it has been reported that clinicians tended to be more
conservative when determining the cardiovascular variant
classification compared with laboratories (20). Therefore, further experiments are
required to verify the pathogenic mechanisms of the variants.
In the present study, it was attempted to amplify
the genomic DNA fragments using PCR and RT-PCR analysis of mRNA was
performed to verify the effect of c.1437C>T (p.Ser479=) and
c.632+4 A>G variants. As only the peripheral blood of the
patients and was obtained the expression levels of CAPN3 in the
blood were low, the amplification was not successful. In addition,
the disease progression of all patients was studied and it was
attempted to obtain their previous medical records. However, due to
disrepair and poor preservation, the results are not fully
available.
The updated criteria for the diagnosis of LGMD
include clinical features of progressive muscle weakness, elevated
serum CK levels, the presence of degenerative changes during muscle
imaging, the presence of dystrophic changes in a muscle biopsy and
genetic inheritance (4). In the
clinic, MRI and muscle biopsies are frequently used to diagnose
muscle dystrophy; however, numerous types of LGMD do not exhibit
specific pathological diagnostic markers or typical clinical
features in the early stages of the disease. Furthermore, western
blot analysis of muscle biopsies indicated that ~20% of patients
had normal CAPN3 protein expression levels (21). In fact, the autocatalytic function
of the proteins was lost. In addition, the European Neuromuscular
Center proposed a new LGMD classification in 2018, which
highlighted the mode of inheritance. The new proposed nomenclature
of LGMD2A is ‘LGMD R1 calpain3-related’, in which the letter ‘R’
means recessive and the number ‘1’ means the order of discovery
(4). Therefore, genetic testing may
become important in preventing invasive testing. In the present
study, next-generation sequencing served a critical role in the
diagnosis of LGMD2A. The six patients all carried complex
heterozygous mutations in the CAPN3 gene and there were no
patients in the next generation, which was consistent with the
autosomal recessive inheritance pattern. WES is a widely used
strategy to detect variants in coding regions and classic splice
sites, and to identify new genes and variants. Considering the
clinical and genetic heterogeneity of LGMD, WES represents a rapid,
cost-effective and accurate method for the diagnosis of the disease
and makes it possible to diagnose both common and rare cases. It
has been reported that the use of WES successfully corrected the
misdiagnosis from non-4q facioscapulohumeral muscular dystrophy to
LGMD2A (22). In fact, with the use
of WES, the variant detection rate has increased from 35 to 45%
(23). As the pathogenesis of
LGMD2A remains unclear, effective treatments currently do not
exist. At present, the major treatments are rehabilitation
treatment, drug therapy and the prevention of complications, which
are aimed at improving the patients' quality of life (14). It is with regret that none of the
patients in this study received good treatment due to the ambiguous
diagnosis prior. In the future, high-throughput technologies will
not only provide us with an accurate diagnosis but also help to
determine the pathological mechanisms of muscle weakness and novel
targets for treatment.
In conclusion, the present study reported four
variants, including two known ‘pathogenic’ and two ‘likely
pathogenic’ variants of CAPN3. According to the analysis of the
genetic patterns in two large Chinese families, clinical evidence
was provided for the classification of the variants. In addition,
the present study emphasized the importance of WES in the diagnosis
and determining the association between the phenotype and genotype
in LGMD2A.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 81771589), the Key
Project of Tianjin Health Care Professionals (grant no. 16KG166)
and the Program of Tianjin Science and Technology Plan (grant no.
18ZXDBSY00170).
Availability of data and materials
The variant c.1437C>T (p.Ser479=) was submitted
to the ClinVar database (submission ID: SUB8078779). The datasets
used and/or analyzed during the current study are available from
the corresponding author on reasonable request.
Authors' contributions
JZ and XX participated in the conception of the
study and writing of the manuscript. JZ, XX, XZ and XW performed
the experiments. XW, JS and CC analyzed the experimental results.
CC revised the manuscript and submitted the manuscript. All of the
authors read and approved the final manuscript.
Ethics approval and consent to
participate
Written informed consent was obtained from all
family members and the study was approved by the ethics committee
of Tianjin Children's Hospital (Tianjin, China).
Patient consent for publication
Written informed consent of the patients were
obtained for publication.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bushby KM: Diagnostic criteria for the
limb-girdle muscular dystrophies: Report of the ENMC consortium on
limb-girdle dystrophies. Neuromuscul Disord. 5:71–74.
1995.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Richard I, Hogrel JY, Stockholm D, Payan
CA, Fougerousse F, Calpainopathy Study Group, Eymard B, Mignard C,
Lopez de Munain A and Fardeau M: Urtizberea JALopez Fardeau
Urtizberea. Natural history of LGMD2A for delineating outcome
measures in clinical trials. Ann Clin Transl Neurol. 3:248–265.
2016.PubMed/NCBI View
Article : Google Scholar
|
|
3
|
Vissing J, Barresi R, Witting N, Van
Ghelue M, Gammelgaard L, Bindoff LA, Straub V, Lochmüller H, Hudson
J, Wahl CM, et al: A heterozygous 21-bp deletion in CAPN3 causes
dominantly inherited limb girdle muscular dystrophy. Brain.
139:2154–2163. 2016.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Straub V, Murphy A and Udd B: LGMD
workshop study group. 229th ENMC international workshop: Limb
girdle muscular dystrophies-nomenclature and reformed
classification Naarden, the Netherlands, 17-19 march 2017.
Neuromuscul Disord. 28:702–710. 2018.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Wang CH, Liang WC, Minami N, Nishino I and
Jong YJ: Limb-girdle muscular dystrophy type 2A with mutation in
CAPN3: The first report in Taiwan. Pediatr Neonatol. 56:62–65.
2015.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Martinez-Thompson JM, Moore SA and
Liewluck T: A novel CAPN3 mutation in late-onset limb-girdle
muscular dystrophy with early respiratory insufficiency. J Clin
Neurosci. 53:229–231. 2018.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Mori-Yoshimura M, Segawa K, Minami N, Oya
Y, Komaki H, Nonaka I, Nishino I and Murata M: Cardiopulmonary
dysfunction in patients with limb-girdle muscular dystrophy 2A.
Muscle Nerve. 55:465–469. 2017.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Okere A, Reddy SS, Gupta S and Shinnar M:
A cardiomyopathy in a patient with limb girdle muscular dystrophy
type 2A. Circ Heart Fail. 6:e12–e13. 2013.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Yalvac ME, Amornvit J, Braganza C, Chen L,
Hussain SA, Shontz KM, Montgomery CL, Flanigan KM, Lewis S and
Sahenk Z: Impaired regeneration in calpain-3 null muscle is
associated with perturbations in mTORC1 signaling and defective
mitochondrial biogenesis. Skelet Muscle. 7(27)2017.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Sorimachi H, Hata S and Ono Y: Calpain
chronicle-an enzyme family under multidisciplinary
characterization. Proc Jpn Acad Ser B Phys Biol Sci. 87:287–327.
2011.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Sorimachi H, Hata S and Ono Y: Impact of
genetic insights into calpain biology. J Biochem. 150:23–37.
2011.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Rychlik W: OLIGO 7 primer analysis
software. Methods Mol Biol. 402:35–60. 2007.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American college
of medical genetics and genomics and the association for molecular
pathology. Genet Med. 17:405–424. 2015.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Angelini C, Giaretta L and Marozzo R: An
update on diagnostic options and considerations in limb-girdle
dystrophies. Expert Rev Neurother. 18:693–703. 2018.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Anthony K, Cirak S, Torelli S, Tasca G,
Feng L, Arechavala-Gomeza V, Armaroli A, Guglieri M, Straathof CS,
Verschuuren JJ, et al: Dystrophin quantification and clinical
correlations in Becker muscular dystrophy: Implications for
clinical trials. Brain. 134:3547–3559. 2011.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Salem IH, Hsairi I, Mezghani N, Kenoun H,
Triki C and Fakhfakh F: CAPN3 mRNA processing alteration caused by
splicing mutation associated with novel genomic rearrangement of
Alu elements. J Hum Genet. 57:92–100. 2012.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Hunt RC, Simhadri VL, Iandoli M, Sauna ZE
and Kimchi-Sarfaty C: Exposing synonymous mutations. Trends Genet.
30:308–321. 2014.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Chamary JV, Parmley JL and Hurst LD:
Hearing silence: Non-neutral evolution at synonymous sites in
mammals. Nat Rev Genet. 7:98–108. 2006.PubMed/NCBI View
Article : Google Scholar
|
|
19
|
Blázquez L, Azpitarte M, Sáenz A,
Goicoechea M, Otaegui D, Ferrer X, Illa I, Gutierrez-Rivas E,
Vilchez JJ and López de Munain A: Characterization of novel CAPN3
isoforms in white blood cells: An alternative approach for
limb-girdle muscular dystrophy 2A diagnosis. Neurogenetics.
9:173–182. 2008.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Bland A, Harrington EA, Dunn K, Pariani M,
Platt J, Grove ME and Caleshu C: Clinically impactful differences
in variant interpretation between clinicians and testing
laboratories: A single-center experience. Genet Med. 20:369–373.
2018.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Fanin M, Nascimbeni AC, Fulizio L,
Trevisan CP, Meznaric-Petrusa M and Angelini C: Loss of calpain-3
autocatalytic activity in LGMD2A patients with normal protein
expression. Am J Pathol. 163:1929–1936. 2003.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Leidenroth A, Sorte HS, Gilfillan G,
Ehrlich M, Lyle R and Hewitt JE: Diagnosis by sequencing:
Correction of misdiagnosis from FSHD2 to LGMD2A by whole-exome
analysis. Eur J Hum Genet. 20:999–1003. 2012.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Angelini C: Neuromuscular disease.
Diagnosis and discovery in limb-girdle muscular dystrophy. Nat Rev
Neurol. 12:6–8. 2016.PubMed/NCBI View Article : Google Scholar
|