Introduction
Rheumatoid arthritis (RA) is an autoimmune disease
of the human body that is characterized by progressive destruction
of the peripheral joint and its surrounding tissues (1). The global incidence of RA has been
estimated to be 0.5-1% (2). The
pathogenesis of RA is complex and may be caused by unknown antigens
that are sensitive to certain inherited factors. Genetic,
epigenetic and environmental factors may contribute to disease
susceptibility and progression. The factors (that require
confirmation), such as viruses, sex hormones and neurodevelopmental
state, are closely related (3). Due
to its numerous complications and high disability rate, RA has a
significant impact on affected individuals, their families and
society, and places an emotional and financial burden on them
(4). Therefore, it is of great
significance to explore the pathogenesis of RA and discover
biomarkers and therapeutic targets for reducing the disability rate
of RA and improving the quality of life of patients.
Long non-coding RNAs (lncRNAs) are >200
nucleotides in length and account for 98% of the ncRNA in the
transcribed human genome (5).
Certain studies have indicated that lncRNAs have an indispensable
role in the pathogenesis of RA (6,7). For
instance, a novel type of lncRNA, C5T1lncRNA, transcribed from a
region between the TNF receptor-associated factor 1 (TRAF1) and
TRAF1-complement component 5 (TRAF1-C5) genes, has been reported to
be mainly expressed in the nucleus. A positive correlation between
C5T1lncRNA and the expression of TRAF1-C5 was identified in
synovial fibroblasts and peripheral blood mononuclear cells (PBMCs)
of patients with RA. C5T1lncRNA is able to participate in the
pathogenesis of RA by regulating the transcription of the TRAF1-C5
gene in the same genomic region (8).
Although the role of lncRNAs in RA has been
increasingly investigated, the current understanding of the
function and regulatory mechanisms of lncRNAs in the development
and progression of RA remains limited. In particular, the
expression of lncRNAs and mRNAs in PBMCs of patients with RA and
the relationship between lncRNAs and clinical indexes remain to be
determined.
In the present study, after comparing 3 RA samples
with 3 negative control (NC) samples to characterize the lncRNA
expression profile in PBMCs, 10 RA samples and 10 NC samples were
selected to evaluate the expression of lncRNA by reverse
transcription-quantitative (RT-q)PCR. The correlations between the
lncRNAs and the RA disease activity index, disease activity score
(DAS) including a 28-joint count rheumatoid arthritis DAS (DAS-28)
and the 36-item short-form survey (SF-36) score (9,10),
were analyzed. The results indicated that differentially expressed
lncRNAs may be involved in the pathogenesis of RA, suggesting that
they may have potential as novel biomarkers for the diagnosis and
prognosis of RA. In the future, experiments with a larger sample
size will be required to verify the present results and to clarify
the regulatory mechanisms of the roles of lncRNAs in patients with
RA.
Materials and methods
Patients and biochemical
measurements
A total of 10 patients with RA were selected from
the Department of Rheumatology and Immunology, the First Affiliated
Hospital of Anhui University of Chinese Medicine (FAHAUCM; Hefei,
China) between June 2019 and July 2019. The cohort comprised 9
females and 1 male with a mean age of 43.7 years (range, 27 to 68
years). Furthermore, 10 normal controls (NCs) were recruited, who
were age and sex-matched with the case group and who received a
routine physical examination at the Department of Health of the
FAHAUCM. All patients with RA fulfilled the new classification
criteria of RA proposed by the American Rheumatology Society and
the European Union against Rheumatism in 2010(11). Patients who had serious heart, liver
or kidney diseases, mental disorders and pregnant or lactating
females were excluded. Informed consent was obtained from all
participants, including the RA group and NC group, prior to
initiation of the study and it was approved by the Ethics Committee
of the FAHAUCM. It was performed in compliance with the
recommendations of the Declaration of Helsinki.
For patients with RA, disease activity was
determined using the disease activity SF-36 scale and the DAS-28
scoring system based on a questionnaire survey (10). This provided an absolute number
reflecting disease activity from four items in DAS-28, including the
number of arthrocele and tender joints, the visual analogue scale
and the erythrocyte sedimentation rate (ESR) in the first hour
(12). ESR, high-sensitivity
C-reactive protein (CRP), rheumatoid factor (RF), anti-cyclic
citrullinated peptide antibody, immunoglobulin A (IgA), IgG, IgM,
C3 and C4 were measured enzymatically using an autoanalyzer
(Hitachi 747; Hitachi). Patients with values of >8 mg/ml for CRP
and RF >20 IU/ml were considered positive for RA.
PBMC preparation and total RNA
extraction
From each donor, 5-ml blood samples collected
immediately were added to 7.5 ml Ficoll-Paque plus (Cytiva) and
centrifuged at 1,2000 g for 15 min (2-8˚C). It has been proposed
that the intermediate flocs should be washed and collected twice
with 9 ml PBD. The intermediate flocs were collected twice by
centrifugation at 7,500 gx5 min (2-8˚C). Subsequently, total RNA
was extracted from the freshly obtained PBMCs using TRIzol reagent
(Takara Bio, Inc.) according to the manufacturer's protocol.
lncRNA and mRNA sequencing (seq)
The quality of all participants' RNA (including
participants who are sequenced first and participants who are
verified later) were evaluated by a nanometer photometer
spectrophotometer (IMPLEN). The Qubit RNA Assay Kit with the Qubit
2.0 Fluorometer (Thermo Fisher Scientific, Inc.) and an Agilent
Bioanalyzer 2100 (Agilent Technologies, Inc.) were used to
determine the RNA concentration and examine the RNA integrity,
respectively. Subsequently, the RNA library was constructed using a
total amount of 3 µg of RNA each sample, and its RNA integrity
number was >7.0. Ribosomal RNA (rRNA) was removed by the
Epicentre Ribo-zero rRNA Removal kit (Epicentre; Illumina, Inc.)
according to the manufacturer's protocol. Afterwards, the
strand-specific sequencing libraries was obtained by the New
England Biolabs (NEB) Next Ultra Directional RNA Library Prep Kit
for Illumina (NEB) using the dUTP method. The RNA-seq assay was
accomplished on an Illumina Hiseq 2000 platform (Illumina, Inc.)
and 100 bp paired-end reads were obtained. The preparation of the
total transcriptome libraries was completed by YuXi Bioinformatics
Corp.
Verification of significantly
upregulated lncRNAs
PBMCs were isolated immediately after 5-ml blood
samples were collected from each donor. PBMCs from 10 patients with
RA and 10 NCs were used for validation of six lncRNAs by RT-qPCR.
Total RNA was obtained from the PBMCs with TRIzol (Thermo Fisher
Scientific, Inc.) and was used for synthesis of complementary DNA
with the ReverTraAc real-time qPCR kit (Takara Bio, Inc.) (13). The amplification conditions were as
follows: Pre-denaturation at 95˚C for 10 min; denaturation at 95˚C
for 15 sec, annealing at 60˚C for 30 sec, and extension for 120
sec. A total of 25 cycles and 30 cycles were used to amplify the
500 bp fragment (14) The primer
sequences are provided in Table I.
Divergent primer design refers to Panda and Gorospe. To synthesize
cDNA from total RNA, Prime-Script Master Mix (Takara Bio, Inc.) was
used (60˚C for 30 min). The relative expression of lncRNA was
subsequently detected using the TB Green Premix Ex Taq II (Tli
RNaseH Plus; Takara Bio, Inc.) with β-actin as an internal control
(15). The relative expression
levels were quantified from three independent experiments by the
2-∆∆Cq method (16).
 | Table ISpecific primers used for quantitative
PCR analysis. |
Table I
Specific primers used for quantitative
PCR analysis.
| Gene name | Sequence (5'-3') | Product length
(bp) |
|---|
| LINC01504 |
F:TTGGCTAACGGAGTTTTGCT | 146 |
| |
R:CTTCTGAGGCCTGGATCTTG | |
| LINC00968 |
F:GCCCAGTTGACAGGAAATGT | 182 |
| |
R:TTGGTTCTCAATGGGATGGT | |
| FAM95B1 |
F:GGAGCTCAGTGCCCTCATAG | 141 |
| |
R:GCTCCAGGATGATGGTGTCT | |
| MIR503HG |
F:CCCCCAACAAAGGAACACTA | 142 |
| |
R:ACTTGGGTGGTTTTCAATGC | |
| LINC00304 |
F:CCGTCCAAGAGCAAAGCTAC | 143 |
| |
R:GGCATCAGGCAAAATCAAGT | |
| LINC01146 |
F:ATTCAGCCAACCAACTGAGG | 147 |
| |
R:TCACAGGTTCTGTGGGTCAA | |
| GAPDH |
F:GGAGCGAGATCCCTCCAAAAT | 205 |
| |
R:GGCTGTTGTCATACTTCTCATGG | |
Functional group analysis
Analysis of the biological functions and signaling
pathways of the abnormally expressed mRNAs was performed by
utilizing the Gene Ontology (GO; www.genontology.org) and Kyoto Encyclopedia of Genes
and Genomes (KEGG; www.genome.ad.jp/KEGG) databases (17,18).
P<0.05 was considered to indicate a statistically significant
difference. The top 20 significantly differentially expressed genes
were subjected to GO functional enrichment analysis. On the
abscissa, the functional terms in the three basic categories of GO
(biological process, cellular component and molecular function)
were presented. The enrichment of KEGG was calculated in a similar
manner as the GO analysis.
Statistical analysis
The statistical significance of differences was
conveniently estimated by a t-test of the RNA-seq data. lncRNAs
(up- or downregulated) with fold changes (FC) ≥2 and P<0.05 were
selected as being significantly differentially expressed and the
false discovery rate was calculated to correct the P-value. Values
are expressed as the mean ± standard deviation. The groups were
compared to evaluate the statistical significance using the
Mann-Whitney U test, Student's t-test, Wilcoxon signed-rank test or
Chi-square test, as appropriate. GraphPad Prism 7.0 (GraphPad
Software, Inc.) and SPSS (version 22.0; IBM Corp.) were used to
analyze all statistical data. P<0.05 was considered to indicate
statistical significance.
Results
Clinical and biochemical features of
the included individuals
As presented in Table
II, there were no obvious differences between the patients with
RA and healthy subjects in terms of sex and age. The baseline
parameters of the two groups were consistent and comparable.
| Table IIClinical characteristics of the study
population. |
Table II
Clinical characteristics of the study
population.
| Index | RA (n=10) | Control (n=10) | P-value |
|---|
| Sex (M/F) | 1:9 | 1:9 | 1 |
| Age (years) | 45.9±11.80 | 43.7±7.99 | 0.637 |
| ESR (mm/h) | 38±27.48 | NA | NA |
| Hs-CRP (mg/l) | 23.40±36.40 | NA | NA |
| RF (U/ml) | 109.48±82.34 | NA | NA |
| Anti-CCP
(U/ml) | 164.75±226.34 | NA | NA |
| IgA (g/l) | 2.53±0.57 | NA | NA |
| IgG (g/l) | 13.88±3.40 | NA | NA |
| IgM (g/l) | 1.48±0.61 | NA | NA |
| C3 (g/l) | 1.23±0.27 | NA | NA |
| C4 (g/l) | 0.34±0.23 | NA | NA |
| DAS-28 score | 6.80±0.98 | NA | NA |
| VAS score | 6.95±0.94 | NA | NA |
| SAS score | 56.4±4.88 | NA | NA |
| SDS score | 56.6±6.07 | NA | NA |
| SF-36 score | 1302.1±14.99 | NA | NA |
lncRNA expression profiles of patients
with RA
In the present study, 231 upregulated and 110
downregulated lncRNAs were identified as differentially expressed
in patients with RA compared with the controls. Information on the
top 6 up- or downregulated lncRNAs is provided in Table III.
| Table IIIInformation on the top six up- or
down-regulated lncRNAs in patients with rheumatoid arthritis
compared with healthy controls. |
Table III
Information on the top six up- or
down-regulated lncRNAs in patients with rheumatoid arthritis
compared with healthy controls.
| lncRNAs | P-value | Fold change | Direction of
regulation | Gene symbol |
|---|
|
ENSG00000246430 | 0.001366 | 2.747691448 | Up | LINC00968 |
|
ENSG00000223749 | 0.002933 | 2.532423677 | Up | MIR503HG |
|
ENSG00000225434 | 0.000301 | -3.210084216 | Down | LINC01504 |
|
ENSG00000223839 | 0.002506 | -2.536755148 | Down | FAM95B1 |
|
ENSG00000180422 | 0.003974 | -2.794683328 | Down | LINC00304 |
|
ENSG00000258867 | 0.004095 | -2.478197951 | Down | LINC01146 |
Analysis of differentially expressed
lncRNAs and mRNAs
From the lncRNA expression profiles, differentially
expressed lncRNAs between the RA group and the NC group were
identified, consisting of 231 upregulated and 110 downregulated
lncRNAs, according to the filtering criteria of P<0.05 and FC
≥2. Graphical presentations of the differentially expressed lncRNAs
are provided in Fig. 1. Scatter
plots and volcano plots were used to display the lncRNAs based on
their expression levels compared between samples (Fig. 1A and C). In order to further reveal the
differences of the lncRNAs between samples, a hierarchical
clustering analysis was applied, as presented in Fig. 1E, and it was indicated that there
was a general difference between patients and controls in the
lncRNAs presented.
In addition, 7,895 differentially expressed mRNAs
between the RA and NC groups were identified, which consisted of
4,916 upregulated mRNAs and 2,934 downregulated mRNAs according to
the filtering criteria of P<0.05 and fold change ≥1.5. Scatter
plot and volcano plot analysis were performed to represent the
differentially expressed mRNAs between the two groups, as depicted
in Fig. 1B and D. Their distinct expression patterns were
also displayed in a hierarchical clustering analysis as presented
in Fig. 1F.
GO analysis of the significantly differentially
expressed mRNAs indicated that organelle organization, protein
modification by small protein conjugation, positive regulation of
cytoplasmic mRNA processing, intracellular, intracellular part,
intracellular organelle, organelle nucleoplasm and intracellular
membrane-bounded organelle were significant terms (Fig. 2). The KEGG analysis of these
significantly differentially expressed mRNAs suggested that the
cyclic AMP signaling pathway, colorectal cancer, Epstein-Barr virus
infection, ErbB signaling pathway, influenza A, proteoglycans in
cancer Ras signaling pathway, renal cell carcinoma, sphingolipid
signaling pathway and TNF signaling pathway were significant
pathways (Fig. S1). Furthermore,
according to the KEGG analysis of the significantly differentially
expressed lncRNAs, the IL-17 signaling pathway, TNF signaling
pathway and intestinal immune network for IgA production were
significant (Table SI).
Finally, to identify interactions between mRNAs and
lncRNAs, gene co-expression networks were constructed, which were
established according to the normalized signal intensity of unit
genes. Data were normalized by using the median gene expression
value of all transcripts expressed from the same coding gene,
without any additional modification of the lncRNA expression value.
The differentially expressed lncRNAs and mRNAs were screened from
the observed list. For each analyzed gene, the Pearson correlation
was calculated and lncRNA-mRNA pairs with significant correlations
were selected in order to construct the network. Cytoscape was used
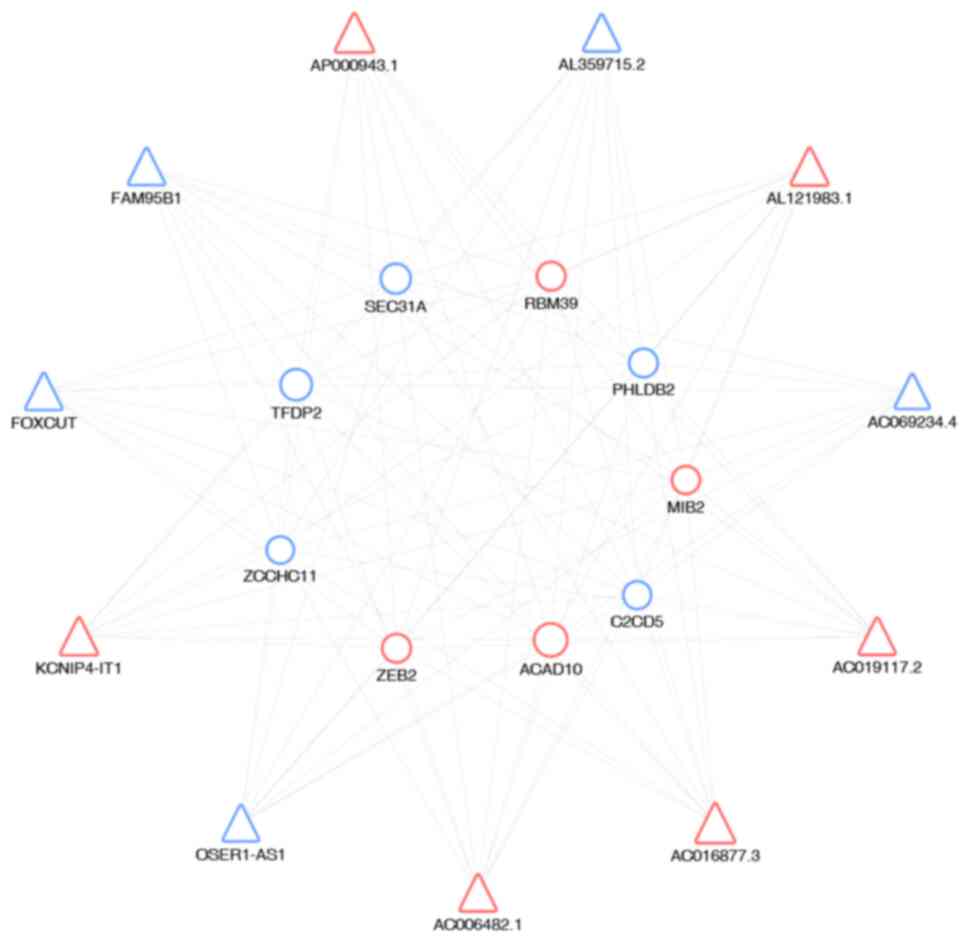
to draw the co-expression networks. In the network (Fig. 3), triangles represent the lncRNAs
and circles the mRNAs. Red and blue represent up- and downregulated
RNAs, respectively. The size of the triangles and circles
represents fold changes of lncRNAs and mRNAs with larger sizes
indicating higher fold changes. The results revealed a complex
lncRNA target network that consisted of 99 matched lncRNA-mRNA
pairs. A detailed co-expression network of lncRNAs and mRNAs is
provided in Fig. S2.
Validation of differentially expressed
lncRNAs
The top six up- or downregulated lncRNAs were
selected. Of these, four were significant, including an upregulated
lncRNA (MIR503HG) and three downregulated lncRNAs (LINC01504,
FAM95B1 and LINC01146). Differentially expressed lncRNAs in
patients with RA determined by RNA-seq were then verified by
RT-qPCR analysis (Fig. 4). The
expression of LNC01504, FAM95B1 and LINC00304 in the control group
was higher than that in the RA group and the expression of MIR503HG
in the RA group was higher than that in the control group. Spearman
tests of the correlation of clinical variables with the lncRNAs in
PBMCs from patients with RA were then performed (Fig. 5). According to the results, there
was a strong positive correlation of FAM95B1 with IgG (r=0.9237,
P<0.05) and C4 (r=0.4869, P<0.05), of LINC00304 with IgG
(r=0.6057, P<0.05) and arthrocele (r=0.5763, P<0.05), of
MIR503HG with joint tenderness (r=0.6083, P<0.05) and of
LINC01504 with course of the disease (r=0.5326, P<0.05), but a
negative correlation between LINC01504 and RF (r=0.4058,
P<0.05).
Discussion
RA is a chronic autoimmune joint disease and its
persistent inflammation affects bone formation and remodeling,
leading to gradual bone destruction (19). Its incidence is high, its
pathogenesis is not clear and so far, there is no efficient cure.
At present, clinical diagnosis is also difficult. The commonly used
imaging and serological tests lack specificity (20). Therefore, the search for specific
and early diagnostic molecular markers is of great significance for
the early diagnosis and treatment of RA.
It was suggested that during the occurrence and
development of RA, lncRNA transcriptional regulation is highly
specific, which may be an important feature for the independent
prediction of biological indicators (6).
TNF receptor-associated factor 1 (TRAF1) and
TRAF1-complement component 5 (TRAF1-C5) are susceptibility genes
for the pathogenesis of RA. The level of TRAF1-C5 in the joints of
patients with RA increases significantly in the acute phase. On the
other hand, the development of arthritis was delayed in an
arthritis model of TRAF1-C5-deficient mice. It was indicated that a
novel type of lncRNA, C5T1lncRNA, was mainly expressed in the
nucleus, transcribed from the regions between the TRAF1 and
TRAF1-C5 genes in RA patients. There was a positive correlation
between C5T1lncRNA and the expression of TRAF1-C5 in synovial
fibroblasts and PBMCs in patients with RA. C5T1lncRNA serves a role
in the pathogenesis of RA by regulating the transcription of the
TRAF1-C5 gene located in the same genomic region (8).
With the completion of the human genome project, it
has been indicated that the number of total protein-encoding genes
in humans is <20,000(21).
Non-coding RNAs refer to a group of RNAs that are not translated
into proteins, including small nuclear RNAs, small nucleolar RNAs,
microRNAs (miRNAs) and lncRNAs (22). These non-coding RNAs manipulate gene
expression at the epigenetic, transcription and
post-transcriptional level and they also participate in almost all
physiologic and pathologic processes (23-25).
lncRNAs influence gene expression at multiple levels
and in multiple ways (26). lncRNAs
either have roles as transcription factors directly or combine with
transcription factors to affect their functions, thereby regulating
the expression of associated genes (27). A study revealed that lncRNA Mrhl
mediated meiotic commitment of mouse spermatogonial cells by
regulating Sox8 expression (28).
Furthermore, lncRNAs act as precursors of miRNAs or as competitive
endogenous RNA, which act as molecular sponges to regulate their
target gene expression by altering the quantity of miRNA (29). Therefore, lncRNAs engage in the
crucial regulation of the biology of various cell types. However,
the precise mechanism of action of lncRNAs in RA remains
elusive.
In the present study, high-throughput sequencing was
used to detect the expression of lncRNA and mRNA in PBMCs of
patients with RA and healthy controls. To the best of our
knowledge, the present study was the first to analyze the
correlation between differentially expressed genes and the clinical
laboratory indexes of patients with RA. The results suggested that
certain lncRNAs were strongly associated with clinical laboratory
indexes. First, the selected differentially expressed lncRNAs were
verified by RT-qPCR. A total of two lncRNAs, LINC00968 and
MIR503HG, whose expression was upregulated during chondrogenesis,
and four of those that were downregulated (LINC01504, FAM95B1,
LINC00304 and LINC01146) were selected. Subsequently, the
differentially expressed mRNAs were analyzed by GO enrichment and
KEGG pathway analysis while the differentially expressed lncRNAs
were analyzed by KEGG pathway analysis. The results indicated that
for patients with RA, significant changes in their lncRNA
expression profile in PBMCs may be related to inflammation. These
pathways are related to signaling pathways such as TGF, TNF, EBRr
and cAMP, which have been widely confirmed to have an important
role in the pathogenesis of RA (30-32).
Subsequently, six differentially expressed lncRNAs
were selected for RT-qPCR verification. The expression of
LINC00304, LINC01504 and FAM95B1 in the RA group was significantly
lower than that in the control group. The expression of MIR503HG in
the RA group was significantly higher than that in the control
group and the results were consistent with the sequencing.
Furthermore, correlations between the expression of LINC00304,
LINC01504, FAM95B1 and MIR503HG with biochemical measurements were
observed. The results suggested that the levels of FAM95B1 were
positively associated with IgG and C4, while the levels of
LINC00304 were positively associated with IgG and arthrocele, and
there was a positive correlation between MIR503HG and joint
tenderness, LINC01504 and course of disease, but a negative
correlation between LINC01504 and RF. These results revealed that
LINC00304, LINC01504, FAM95B1 and MIR503HG may have critical roles
in the pathogenic mechanism of RA.
In RA, as a chronic disease, the presentation and
course of the disease differ among individual patients. In recent
studies, gene detection in RA has been frequently discussed, but
the results were rarely combined with clinical indicators and the
patients' quality of life (33,34).
In conclusion, the present study may provide a
reference for development of diagnostic tools. However, due to the
limitation of the small number of samples, there are still certain
deficits and in future studies, a larger cohort will be used to
confirm the present results.
Supplementary Material
KEGG pathways of the differentially
expressed mRNAs. The abscissa displays the enrichment factor and
the ordinate presents the pathway terms with the highest degree of
enrichment (the 20 most enriched ones are selected for display).
P<0.05 was considered to indicate a statistically significant
difference. The colour indicates the P-value with red indicating
the most significant enrichment. The size of the dots indicates the
number of differentially expressed genes accumulated in the term.
KEGG, Kyoto Encyclopedia of Genes and Genomes.
lncRNA-mRNA co-expression network.
Construction of the lncRNA-mRNA co-expression network. Triangles
represent lncRNAs and circles mRNAs. Red and blue represent up. and
downregulated RNAs, respectively. The size of the triangles and
circles represents the fold changes of the RNAs. lncRNA, long
non-coding RNA.
Enriched Kyoto Encyclopedia of Genes
and Genomes pathways of differentially expressed long non-coding
RNAs.
Acknowledgements
Not applicable.
Funding
The present study was funded by the National Nature
Fund Program (grant no. 81973655), The Anhui Key Research and
Development Program Foreign Science and Technology Cooperation
Project (grant no. 201904b11020011), the Ministry of Science and
Technology National Key Research and Development Program Chinese
Medicine Modernization Research Key Project (grant no.
2018YFC1705204), the Anhui Provincial Quality Engineering Teaching
and Research Project (grant no. 2018jyxm1068), the Key Research and
Development Plan Project of Anhui Province (grant no.
201904a07020004), the Anhui Provincial Laboratory of Applied Basis
and Development of Internal Medicine of Modern Traditional Chinese
Medicine (grant no. 2016080503B041) and the 12th Batch of the ‘115’
Innovation Team of Anhui Province [Anhui Talent Office (2019) No.
1], the Key Laboratory opening Project of Xin'an Ministry of
Medical Education (2020xayx08).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JL, HJ, LW, YS, LX, PZ and DH contributed to the
study design. YL and JW contributed to data analysis, wrote the
first draft and revised the manuscript. YS, YL, YZ, BB and GS
contributed to the specimen and data collection. JL and HJ were the
supervisors of the project and contributed to the revision of the
manuscript. All authors reviewed and approved the final
manuscript.
Ethics approval and consent to
participate
The study complied with the Declaration of Helsinki
and was approved by the Institutional Review Board Ethics Committee
of The First Affiliated Hospital of Anhui University of Chinese
Medicine. Written informed consent was obtained from each
patient.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
McInnes IB and Schett G: Pathogenetic
insights from the treatment of rheumatoid arthritis. Lancet.
389:2328–2337. 2017.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Catrina AI, Svensson CI, Malmstrom V,
Schett G and Klareskog L: Mechanisms leading from systemic
autoimmunity to joint-specific disease in rheumatoid arthritis. Nat
Rev Rheumatol. 13:79–86. 2017.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Michels AW and Eisenbarth GS: Immunologic
endocrine disorders. J Allergy Clin Immunol. 125 (Suppl
2):S226–S237. 2010.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Sharif K, Sharif A, Jumah F, Oskouian R
and Tubbs RS: Rheumatoid arthritis in review: Clinical, anatomical,
cellular and molecular points of view. Clin Anat. 31:216–223.
2018.PubMed/NCBI View
Article : Google Scholar
|
|
5
|
Kowalczyk MS, Higgs DR and Gingeras TR:
Molecular biology: RNA discrimination. Nature. 482:310–311.
2012.PubMed/NCBI View
Article : Google Scholar
|
|
6
|
Muller N, Doring F, Klapper M, Neumann K,
Schulte DM, Turk K, Schröder JO, Zeuner RA, Freitag-Wolf S,
Schreiber S and Laudes M: Interleukin-6 and tumour necrosis
factor-alpha differentially regulate lincRNA transcripts in cells
of the innate immune system in vivo in human subjects with
rheumatoid arthritis. Cytokine. 68:65–68. 2014.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Luo Q, Xu C, Li X, Zeng L, Ye J, Guo Y,
Huang Z and Li J: Comprehensive analysis of long non-coding RNA and
mRNA expression profiles in rheumatoid arthritis. Exp Ther Med.
14:5965–5973. 2017.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Messemaker TC, Frank-Bertoncelj M, Marques
RB, Adriaans A, Bakker AM, Daha N, Gay S, Huizinga TW, Toes RE,
Mikkers HM and Kurreeman F: A novel long non-coding RNA in the
rheumatoid arthritis risk locus TRAF1-C5 influences C5 mRNA levels.
Genes Immun. 17:85–92. 2016.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Moerman RV, Arends S, Mossel E, Kroeze
FGM, Vissink A and Bootsma H: 10-year follow-up of patients with
rheumatoid arthritis and secondary Sjogren's syndrome or sicca
symptoms in daily clinical practice. Clin Exp Rheumatol. 38 (Suppl
126):S64–S72. 2020.PubMed/NCBI
|
|
10
|
Sul B, Lee KB, Joo YB, Hong BY, Kim JS,
Kim KJ, Park KS, Park YJ and Lim SH: Twelve weeks of strengthening
exercise for patients with rheumatoid arthritis: A prospective
intervention study. J Clin Med. 9(E2792)2020.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Aletaha D, Neogi T, Silman AJ, Funovits J,
Felson DT, Bingham CO III, Birnbaum NS, Burmester GR, Bykerk VP,
Cohen MD, et al: 2010 Rheumatoid arthritis classification criteria:
An American College of Rheumatology/European League Against
Rheumatism collaborative initiative. Arthritis Rheum. 62:2569–2581.
2010.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Ramírez J, Cuervo A, Celis R, Ruiz-Esquide
V, Castellanos-Moreira R, Narváez JA, Gómez-Puerta JA, Pablos JL,
Sanmartí R and Cañete JD: Biomarkers for treatment change and
radiographic progression in patients with rheumatoid arthritis in
remission: A 5 year follow-up study. Rheumatology (Oxford): Jul 12,
2020 (Epub ahead of print). doi:
org/10.1093/rheumatology/keaa258.
|
|
13
|
Wen J, Liu J, Zhang P, Jiang H, Xin L, Wan
L, Sun Y, Huang D, Sun Y, Long Y, et al: RNA-seq reveals the
circular RNA and miRNA expression profile of peripheral blood
mononuclear cells in patients with rheumatoid arthritis. Biosci
Rep. 40(BSR20193160)2020.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Ju H, Zhang L, Mao L, Wu Y, Liu S, Ruan M,
Hu J and Ren G: A comprehensive genome-wide analysis of the long
noncoding RNA expression profile in metastatic lymph nodes of oral
mucosal melanoma. Gene. 675:44–53. 2018.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Yin J, Hu T, Xu L, Li P, Li M, Ye Y and
Pang Z: Circular RNA expression profile in peripheral blood
mononuclear cells from Crohn disease patients. Medicine
(Baltimore). 98(e16072)2019.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Si C, Wang J, Ma W, Hua H, Zhang M, Qian
W, Zhou B and Luo D: Circular RNA expression profile in human
fibroblast premature senescence after repeated ultraviolet B
irradiations revealed by microarray. J Cell Physiol.
234:18156–18168. 2019.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Zhao Z, Bai J, Wu A, Wang Y, Zhang J, Wang
Z, Li Y, Xu J and Li X: Co-lncRNA: Investigating the lncRNA
combinatorial effects in GO annotations and KEGG pathways based on
human RNA-Seq data. Database (Oxford). 2015(bav082)2015.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Yang L, Lyu L, Wu W, Lei D, Tu Y, Xu D,
Feng J and He L: Genome-wide identification of long non-coding RNA
and mRNA profiling using RNA sequencing in subjects with sensitive
skin. Oncotarget. 8:114894–114910. 2017.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Harnden K, Pease C and Jackson A:
Rheumatoid arthritis. BMJ. 352(i387)2016.PubMed/NCBI View
Article : Google Scholar
|
|
20
|
Pincus T and Sokka T: Laboratory tests to
assess patients with rheumatoid arthritis: Advantages and
limitations. Rheum Dis Clin North Am. 35:731–734, vi-vii.
2009.PubMed/NCBI View Article : Google Scholar
|
|
21
|
ENCODE Project Consortium. Birney E,
Stamatoyannopoulos JA, Dutta A, Guigo R, Gingeras TR, Margulies EH,
Weng Z, Snyder M, Dermitzakis ET, et al: Identification and
analysis of functional elements in 1% of the human genome by the
ENCODE pilot project. Nature. 447:799–816. 2007.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Klingenberg M, Matsuda A, Diederichs S and
Patel T: Non-coding RNA in hepatocellular carcinoma: Mechanisms,
biomarkers and therapeutic targets. J Hepatol. 67:603–618.
2017.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Fruci D, Rota R and Gallo A: The Role of
HCMV and HIV-1 MicroRNAs: Processing, and mechanisms of action
during viral infection. Front Microbiol. 8(689)2017.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Sahni A, Hajjari M, Raheb J, Foroughmand
AM and Asgari M: Cloning and over expression of non-coding RNA rprA
in E.coli and its resistance to Kanamycin without osmotic shock.
Bioinformation. 13:21–24. 2017.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Zhang J, Le TD, Liu L and Li J: Inferring
miRNA sponge co-regulation of protein-protein interactions in human
breast cancer. BMC Bioinformatics. 18(243)2017.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Deniz E and Erman B: Long noncoding RNA
(lincRNA), a new paradigm in gene expression control. Funct Integr
Genomics. 17:135–143. 2017.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Luo G, Liu D, Huang C, Wang M, Xiao X,
Zeng F, Wang L and Jiang G: lncRNA GAS5 inhibits cellular
proliferation by targeting P27Kip1. Mol Cancer Res.
15:789–799. 2017.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Kataruka S, Akhade VS, Kayyar B and Rao
MRS: Mrhl long noncoding RNA mediates meiotic commitment of mouse
spermatogonial cells by regulating sox8 expression. Mol Cell Biol.
37:e00632–16. 2017.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Laneve P, Po A, Favia A, Legnini I, Alfano
V, Rea J, Di Carlo V, Bevilacqua V, Miele E, Mastronuzzi A, et al:
The long noncoding RNA linc-NeD125 controls the expression of
medulloblastoma driver genes by microRNA sponge activity.
Oncotarget. 8:31003–31015. 2017.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Cui Y, Yi Q, Sun W, Huang D, Zhang H, Duan
L, Shang H, Wang D and Xiong J: Molecular basis and therapeutic
potential of myostatin on bone formation and metabolism in
orthopedic disease. BioFactors: Sep 30, 2020 (Epub ahead of print).
doi: 10.1002/biof.1675.
|
|
31
|
Cheng Q, Wu H and Du Y: The roles of
small-molecule inflammatory mediators in rheumatoid arthritis.
Scand J Immunol: Oct 8, 2020 (Epub ahead of print). doi:
10.1111/sji.12982.
|
|
32
|
Wu HX, Chen JY, Wang QT, Sun WY, Liu LH,
Zhang LL and Wei W: Expression and function of β-arrestin 2
stimulated by IL-1β in human fibroblast-like synoviocytes and the
effect of paeoniflorin. Int Immunopharmacol. 12:701–706.
2012.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Berner C, Erlacher L, Fenzl KH and Dorner
TE: A cross-sectional study on self-reported physical and mental
health-related quality of life in rheumatoid arthritis and the role
of illness perception. Health Qual Life Outcomes.
16(238)2018.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Al-Herz A, Saleh K, Al-Awadhi A,
Al-Kandari W, Hasan E, Ghanem A, Hussain M, Ali Y, Nahar E, Alenizi
A, et al: Accessibility to biologics and its impact on disease
activity and quality of life in patients with rheumatoid arthritis
in Kuwait. Clin Rheumatol: Oct, 12, 2020 (Epub ahead of print). doi
org/10.1007/s10067-020-05444-2.
|