Introduction
Lung cancer is one of the most life-threatening
malignancies worldwide, accounting for ~20% of all
cancer-associated deaths and with >2 million estimated new cases
in 2018(1). Lung cancer is
classified as small cell lung cancer and non-small cell lung
cancer, of which lung adenocarcinoma (LUAD) and lung squamous cell
carcinoma are the most common subtypes (2). Although significant progress has been
made in diagnosis and treatment during the last few decades
(2), the prognosis of patients with
LUAD remains poor. Therefore, the identification of novel molecules
involved in the progression of LUAD may benefit the development of
early diagnostic methods and/or targeted therapies.
AKT is a common component of multiple signaling
pathways with a variety of downstream effectors (3), thereby integrating different upstream
signals to regulate numerous cellular activities, including protein
synthesis, migration and the cell cycle (3). The mechanistic target of rapamycin
(mTOR) is one of the proteins downstream of AKT, and one of three
core components of mTOR complex 1 (mTORC1). mTORC1 coordinates
cellular proliferation and metabolism with nutrients and growth
factors, as well as inhibits autophagy (4). Hyperactivation of AKT and mTORC1 has
been observed in numerous types of cancer, including LUAD and
colorectal carcinoma (3,4).
The epidermal differentiation complex (EDC) is a
gene cluster located on human chromosome 1q21(5). Protein NICE-3 (also known as
chromosome 1 open reading frame 43) is an EDC member originally
identified from a keratinocyte cDNA library (5), which is expressed in the kidney and 25
other human tissues (6). NICE-3
primarily localizes to the Golgi and mitochondria, and interacts
with plasma membrane proteins (7).
Human histiocytic lymphoma U937 cells with NICE-3-deletion exhibit
a strong defect in bacterial uptake (7), indicating its role as a regulator of
phagocytosis (7). Furthermore,
NICE-3 expression is upregulated in human hepatocellular carcinoma
(HCC) and may contribute to HCC progression by promoting cellular
proliferation, colony formation and the cell cycle (8). However, the detailed mechanisms
underlying these phenomena remain unknown.
To the best of our knowledge, there are currently no
studies on the function of NICE-3 in LUAD. Therefore, the aim of
the current study was to investigate NICE-3 expression in LUAD
tissues and its association with patient prognosis, as well as its
function and associated underlying mechanisms.
Materials and methods
Cell culture and transfection
The human LUAD A549 and H1993 cell lines were
acquired from the Kunming Cell Bank of the Chinese Academy of
Sciences (cat. KCB200434YJ; https://www.kmcellbank.com/index.php?c=content&a=show&id=244)
and the Chinese Tissue Culture Collections (cat. no. CTCC-007-0056;
https://ctcc.online/), respectively. Cells were
cultured in RPMI-1640 medium supplemented with 10% fetal bovine
serum (FBS; both Gibco; Thermo Fisher Scientific, Inc.) and 1%
penicillin/streptomycin, and maintained in a humidified incubator
at 37˚C with 5% CO2.
The sequences of the small interfering RNAs (siRNAs)
targeting NICE-3 were as follows: si-NICE-3-#1,
5'-CCUACGGGAGCCUGGACUUTT-3'; si-NICE-3-#2,
5'-GCUAUGAAACAGCCCGCUATT-3'; and si-NICE-3-#3,
5'-GCUUGUGUCUAAAGGGUAATT-3'. The sequence of the non-targeting
control siRNA (si-control) was 5'-UUCUCCGAACGUGUCACGUTT-3'. All
siRNAs were synthesized by Shanghai GenePharma Co., Ltd. A549 cells
(1.5x105/well) were seeded into six-well plates and
cultured until ~70% confluent. The cells were transfected at 37˚C
for 48 h with 75 pmol NICE-3 or control siRNA using
Lipofectamine® 3000 (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's instructions.
For autophagy assay, A549 and H1993 cells were
transfected with si-NICE-3 or si-control for 48 h before being
treated with the autophagy inhibitor bafilomycin A1 (100 nM;
Targetmol) at room temperature for 1 h, followed by western
blotting assay.
Cell counting and colony formation
assay
At 24 h post-transfection, A549 cells were harvested
and manually counted under an optical microscope. The cells
(6x102/well) were subsequently seeded into a 6-well
plate, and cultured in RPMI 1640 medium (with 10% FBS) in a
humidified incubator at 37˚C for 10-14 days. The culture media were
replaced every 2 days until the end of the experiment. Finally, the
cells were fixed with 4% paraformaldehyde for 30 min at room
temperature (RT), and then stained with 0.1% crystal violet at RT
for 1 h. The plates were dried and scanned using a Epson Perfection
V370 Photo scanner. Cell colonies (>50 cells) were counted
manually.
Cell migration and invasion
assays
The migration and invasion assays were performed as
previously described (9). Transwell
inserts (pore size, 8 µm) were coated with Matrigel at 37˚C for 2
h. A549 cells (3x104) were resuspended in 200 µl
serum-free RPMI medium and seeded into the upper chambers of the
Transwell inserts (Costar; Corning Inc.). A total of 600 µl RPMI
medium supplemented with 20% FBS was added to the lower chambers.
The cells were cultured at 37˚C for 24 h. The Transwell inserts
were removed from the plate and the non-invasive or non-migratory
cells were removed with a cotton swab. The migratory cells were
fixed with 4% paraformaldehyde at room temperature for 15 min and
stained with 0.1% crystal violet at 4˚C overnight. Images of three
randomly selected fields per well were captured using a TH4-200
light microscope (x200 magnification; Olympus Corporation).
Protein preparation and western
blotting
Western blot analysis was conducted as previously
described (10). At 48 h
post-transfection, the cells were harvested and lysed in RIPA lysis
buffer (Beijing Solarbio Science & Technology Co., Ltd.) for 15
min on ice. The supernatants were collected before the protein
concentration was determined using a bicinchoninic acid protein
assay kit (Beyotime Institute of Biotechnology). The samples (30 µg
protein) were resolved via 12 or 15% SDS-PAGE and transferred onto
PVDF membranes, which were then blocked with 5% non-fat milk at RT
for 2 h. The membranes were incubated with primary antibodies at
4˚C overnight, washed three times with TBS-Tween (0.1%) and then
incubated with the secondary antibody at RT for 1 h. The blots were
washed and developed using BeyoECL Plus reagent (cat. no. P0018;
Beyotime Institute of Biotechnology). The following primary
antibodies were used: Anti-LC3B (cat. no. nb600-1384; 1:1,000) and
anti-p62/sequestosome 1 (SQSTM1; cat. no. nbp1-48320; 1:1,000)
purchased from Novus Biologicals, LLC; anti-phosphorylated (p)-AKT
(cat. no. 4060; 1:1,000), anti-AKT (cat. no. 9272; 1:1,000),
anti-p-S6K1 (cat. no. 9204; 1:1,000) and anti-S6K1 (cat. no. 9202;
1:1,000) purchased from Cell Signaling Technology, Inc.; mouse
anti-β-actin antibody (cat. no. TA811000; 1:1,000) purchased from
OriGene Technologies, Inc. The secondary antibodies were as
follows: Horseradish peroxidase (HRP)-conjugated anti-mouse IgG
(cat. no. 7076; Cell Signaling Technology, Inc.; 1:5,000) and
HRP-conjugated anti-rabbit IgG (cat. no. 7074; Cell Signaling
Technology, Inc.; 1:5,000). Densitometric analysis was performed
using ImageJ software (v1.53a; National Institutes of Health).
Flow cytometry
A549 cells were collected 48 h after transfection
and washed three times with pre-cooled PBS. The cells were then
fixed with 70% ethanol overnight at 4˚C, followed by a further
three washes with cold PBS. The cells (2x105) were
stained with the staining solution (20 µg/ml propidium iodide and
200 µg/ml RNase; cat. no. F10797; Thermo Fisher Scientific, Inc.)
and then incubated at RT for 1 h in a dark chamber. The cell cycle
was analyzed by flow cytometry (NovoCyte 2060R; ACEA Biosciences,
Inc.) with Software NovoExpress 1.4.0 (ACEA Biosciences, Inc.).
Analysis of NICE-3 expression and
patient prognosis in LUAD
NICE-3 expression analysis in the LUAD dataset from
The Cancer Genome Atlas (TCGA) was conducted using UALCAN (11). The expression of NICE-3 in lung
adenocarcinoma tissue and normal lung tissue from the GSE31210
microarray dataset (12) on Gene
Expression Omnibus database (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE31210)
was analyzed using GraphPad Prism 5.0 (GraphPad Software, Inc.).
The effects of NICE-3 expression on overall survival (OS), first
progression (FP) and post-progression survival (PPS) were
determined using the Kaplan-Meier Plotter online tool (www.kmplot.com) (13).
The cut-off values were automatically determined by the software.
Log-rank test was used to evaluate the differences between survival
curves. The Cox proportional hazards model in the Kaplan-Meier
Plotter online tool was used for multivariate analysis of the
effects of clinical characteristics and NICE-3 expression on OS and
FP.
Statistical analysis
All statistical analyses were conducted using
GraphPad Prism 8 (GraphPad Software, Inc.). All experiments were
repeated ≥ three times independently. Values are expressed as the
mean ± standard deviation. Unpaired Student's t-test was used for
comparisons between two groups, and ANOVA followed by Tukey's
post-hoc test was used for comparisons among multiple groups.
P<0.05 was considered to indicate a statistically significant
difference.
Results
NICE-3 expression is increased in LUAD
tissues, and high expression is associated with poor survival
outcomes
The UALCAN online tool was used to determine NICE-3
expression in TCGA LUAD dataset (11). The results revealed that NICE-3
expression was significantly increased in LUAD compared with in
normal lung samples (P<0.0001; Fig.
1A). To confirm this finding, the GSE31210 dataset from the
Gene Expression Omnibus database was also analyzed (12). The results also demonstrated that
NICE-3 expression was significantly upregulated in LUAD compared
with in normal lung samples (P<0.01; Fig. 1B), consistent with the results of
TCGA LUAD dataset.
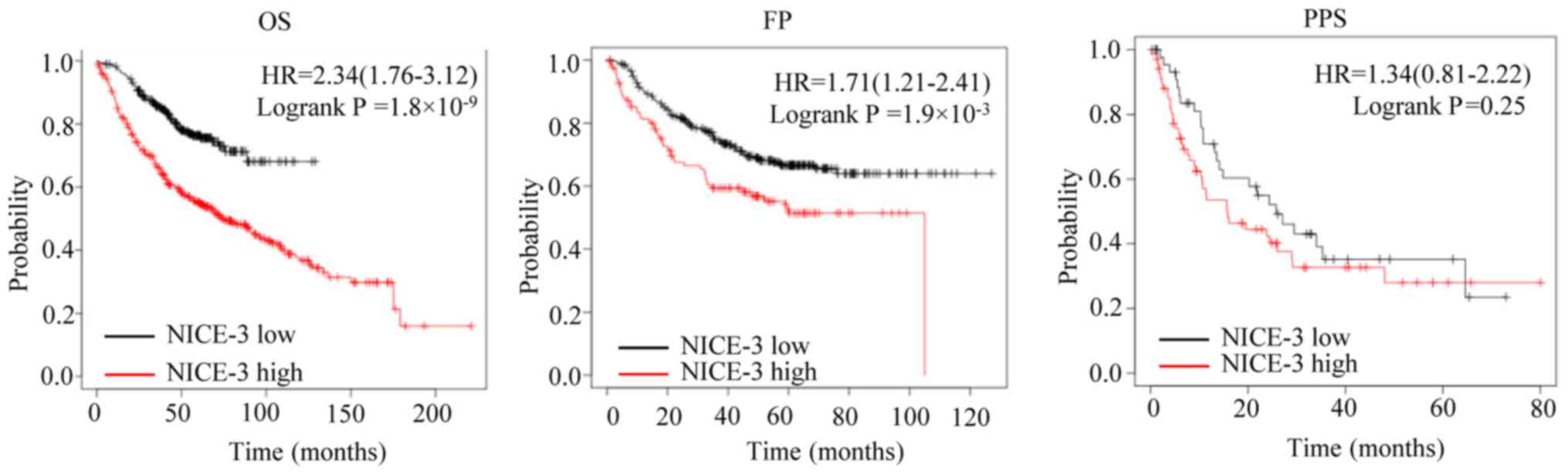
In order to assess the prognostic value of NICE-3 in
patients with LUAD, the associations between NICE-3 expression and
OS, FP and PPS were analyzed using the Kaplan-Meier plotter
(13). The patients were separated
into low and high NICE-3 expression groups according to the cut-off
values determined by the software (177 for OS, 178 for FP and 148
for PPS). The results suggested that high NICE-3 expression was
significantly associated with a poorer OS and FP compared with low
NICE-3 expression, while no significant association with PPS was
observed (Fig. 2), implying that
NICE-3 expression may serve as a predictive indicator of OS and FP.
Furthermore, the results of the multivariate analysis of clinical
characteristics, such as American Joint Committee on Cancer stage
(14), sex and smoking history,
suggested that high NICE-3 expression may be an independent
prognostic indicator for OS only (Table
I). In addition, the results indicated that AJCC stage T
associated significantly with OS and FP (Table I).
 | Table ICox regression multivariate analysis
of NICE-3 expression and OS or FP in patients with lung
adenocarcinoma. |
Table I
Cox regression multivariate analysis
of NICE-3 expression and OS or FP in patients with lung
adenocarcinoma.
| A, OS |
|---|
| Variable | Hazard ratio (95%
CI) | P-value |
|---|
| Sex | 1.39 (0.74-2.61) | 0.3005 |
| Stage | 2.88
(0.48-17.13) | 0.2453 |
| AJCC T stage | 3.08 (1.36-6.98) | 0.0069a |
| AJCC N stage | 0.91 (0.15-5.36) | 0.9178 |
| Smoking history | 0.85 (0.36-1.97) | 0.6988 |
| NICE-3
expression | 0.51 (0.27-0.94) | 0.0305b |
| B, FP |
| Variable | Hazard ratio (95%
CI) | P-value |
| Sex | 1.24 (0.62-2.48) | 0.5460 |
| Stage | 0.93
(0.08-10.32) | 0.9506 |
| AJCC T stage | 4.24
(1.48-12.18) | 0.0073a |
| AJCC N stage | 2.33
(0.22-24.94) | 0.4829 |
| Smoking history | 1.55 (0.69-3.48) | 0.2863 |
| NICE-3
expression | 0.58 (0.29-1.16) | 0.1265 |
NICE-3-knockdown inhibits the
proliferation, cell cycle progression, migration and invasion of
LUAD cells
To determine how NICE-3 negatively impacts patient
prognosis, the LUAD A549 cell line was used to perform in
vitro experiments. A total of three siRNA molecules
specifically targeting NICE-3 (si-NICE-3) were designed, and their
efficacy was confirmed in A549 and H1993 cells transfected with
si-NICE-3 or si-control 48 h after transfection. NICE-3 protein
expression was subsequently detected by western blotting. The blots
demonstrated that si-NICE-3-#3 exerted the strongest inhibitory
effect on NICE-3 protein expression and was thus selected for
subsequent experimentation (Fig.
3A).
To investigate the effects of NICE-3-knockdown on
cellular proliferation, A549 and H1993 cells were transfected with
si-NICE-3 and counted 24 h later. The results revealed that
compared with the control group, NICE-3-knockdown significantly
decreased cell numbers (Fig. 3B),
indicating that cellular proliferation was impaired by
NICE-3-knockdown. A colony formation assay was performed to assess
the effects of NICE-3 on colony formation ability following
NICE-3-knockdown. The results demonstrated that NICE-3-knockdown
significantly compromised colony formation capacity (Fig. 3C), suggesting that NICE-3 may be
involved in cellular survival.
In order to elucidate how cellular proliferation and
survival are influenced by NICE-3, the A549 cell cycle was analyzed
via flow cytometry following NICE-3-knockdown. The results
demonstrated that compared with the si-control, si-NICE-3 induced
G1 phase arrest (Fig.
3D), indicating that NICE-3 positively regulated the cell
cycle, and thus enhanced cellular proliferation and survival. The
H1993 cell cycle was also analyzed via flow cytometry following
NICE-3 knockdown, but no significant effect was observed (data not
shown).
To investigate the role of NICE-3 in cellular
migration and invasion, A549 and H1993 cells were transfected with
si-NICE-3 or si-control prior to Transwell migration and invasion
assays, respectively. The results demonstrated that
NICE-3-knockdown significantly inhibited the migration and invasion
of A549 and H1993 cells (Fig. 4),
suggesting that NICE-3 may be positively associated with the
migration and invasion of LUAD cells.
NICE-3-knockdown promotes autophagy in
LUAD cells
To investigate the potential role of NICE-3 in
autophagy, A549 and H1993 cells were transfected with si-NICE-3 or
si-control for 48 h, and then treated with the autophagy inhibitor
bafilomycin A1 (100 nM) for 1 h. The autophagic marker proteins
LC3B and p62/SQSTM1 were then detected by western blot analysis
(15). The results indicated that
NICE-3-knockdown promoted the conversion of LC3-I to LC3-II, as
well as the degradation of p62/SQSTM1 (Fig. 5A). Following treatment with
bafilomycin A1, LC3-II and p62/SQSTM1 had notably accumulated in
the si-NICE-3 group compared with in the control group (Fig. 5A). These findings suggested that
NICE-3 inhibited A549 and H1993 cell autophagy.
 | Figure 5NICE-3-knockdown induces autophagy by
inhibiting the AKT/mTORC1 signaling pathway. A549 and H1993 cells
were transfected with si-NICE-3 or si-control for 48 h, followed by
bafilomycin treatment for 1 h. LC3 and p62 proteins, as well as AKT
and S6K phosphorylation, were evaluated by western blot analysis.
(A) Conversion of LC3-I to LC3-II (LC3-II/LC3-I ratio as an
indicator) was enhanced, and p62 protein levels were decreased by
si-NICE-3, indicating increased autophagy. Representative western
blots are shown. The bands were quantified using ImageJ and
normalized to the loading control. (B) Phosphorylation of AKT and
S6K were suppressed by NICE-3-knockdown, indicating decreased AKT
and mTORC1 activity. Representative western blots are shown. The
bands were quantified using ImageJ and normalized to the loading
control. *P<005; **P<0.01;
***P<0.001. si, small interfering RNA; mTORC1,
mechanistic target of rapamycin complex 1; Baf, bafilomycin; p,
phosphorylated. |
NICE-3-knockdown inhibits AKT/mTORC1
signaling in LUAD cells
To investigate the underlying mechanisms by which
NICE-3 may regulate autophagy, A549 and H1993 cells were
transfected with si-NICE-3 or si-control for 48 h, and then the
levels of AKT, p-AKT, p70 S6K and p-p70 S6K were detected by
western blotting. The results revealed that NICE-3-knockdown
significantly inhibited the phosphorylation of AKT and p70 S6K
without influencing total AKT and p70 S6K protein levels (Fig. 5B), suggesting that NICE-3 may
function through the AKT/mTORC1 signaling pathway.
Discussion
NICE-3 is a rarely studied molecule, which was first
identified from a keratinocyte cDNA library by Marenholz et
al (5) in 2001. After over a
decade, Wei et al (8)
reported that NICE-3 expression was upregulated in HCC and was
involved in cellular proliferation, colony formation and the cell
cycle. However, the potential role that NICE-3 serves in other
types of cancer remained unclear. By analyzing its expression
levels in two LUAD datasets, the present study revealed that NICE-3
expression was increased in LUAD, and that high expression levels
were associated with poor OS and FP. The current findings indicated
that NICE-3 may be a potential prognostic marker in LUAD.
Excessive proliferation is a typical feature of
malignant tumor growth (16). Since
NICE-3 was found to promote cell cycle progression in HCC (8), the present study hypothesized that
inhibiting NICE-3 expression may inhibit the cell cycle in LUAD
cells. Therefore, the effects of NICE-3 expression were assessed
in vitro using LUAD cells. The results demonstrated that
NICE-3-knockdown in A549 cells inhibited cellular proliferation and
induced cell cycle arrest, consistent with previous observations in
HCC (8). Cellular migration and
invasion are essential features for tumor metastasis (16). The results of the present study
indicated that NICE-3-knockdown inhibited A549 and H1993 cell
migration and invasion, suggesting that NICE-3 may be involved in
tumor metastasis.
Autophagy is a conserved pathway through which
damaged proteins and organelles are degraded to maintain cellular
homeostasis or in response to extracellular cues (17). Previous studies have revealed that
autophagy may function as a tumor-suppressing or -promoting
process, depending on the type of tumor or context (18). Becn1 heterozygous-deficient
mice are prone to the development of liver and lung tumors
(19,20). Basal autophagy is upregulated in
hypoxic tumor regions of cervical cancer where it is essential for
cell survival (21). Wei et
al (15) reported that
autophagy inhibition was associated with increased clonogenic
survival of non-small cell lung cancer cells. Thus, the role of
NICE-3 on autophagy was investigated in the current study,
revealing that NICE-3 may inhibit autophagy.
Although NICE-3 serves an oncogenic role in HCC
(8), the associated underlying
mechanisms remain unknown. The results of the present study
revealed a novel mechanism by which NICE-3-knockdown decreased AKT
and S6K phosphorylation, indicating that NICE-3 positively
regulated AKT and mTORC1 activity in LUAD cells, and thus enhanced
AKT/mTORC1 signaling. Since AKT promotes cellular proliferation,
motility and cell cycle progression (3,22),
NICE-3-knockdown induced cell cycle arrest, attenuated cellular
proliferation and suppressed the migration and invasion of LUAD
cells. To the best of our knowledge, the present study was the
first to reveal the association between NICE-3 expression, the
AKT/mTORC1 signaling pathway and autophagy.
In the present study, in vitro
experimentation was conducted by knocking down NICE-3 expression in
A549 and H1993 cells. Therefore, in order to confirm the present
findings, further experiments, including NICE-3 overexpression and
knockdown, should be conducted in other lung cancer cell lines. In
addition, in vivo LUAD tumor xenograft experiments are
required to confirm the present in vitro findings. Since
autophagy was only examined by detecting LC3-I/II and p62 protein
levels using western blotting, further experiments should be
performed in the future to monitor autophagy using additional
techniques, such as transmission electron microscopy and GFP-LC3
fluorescence microscopy, to confirm the effect of NICE-3 on
autophagy.
In conclusion, the results of the current study
revealed that NICE-3 expression was increased in LUAD tissues, and
that high expression levels were associated with a poor prognosis.
Furthermore, NICE-3 promoted the proliferation, cell cycle
progression, invasion and migration of LUAD cells, as well as
suppressed autophagy. Therefore, NICE-3 may serve an oncogenic role
in LUAD and may be a potential therapeutic target.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by the Hundred Talents
Program of Guangxi, Natural Science Foundation of Guangxi (grant
no. 2020GXNSFAA297209), the Research Enhancement Project for Junior
Faculty in Higher Education Institutes of Guangxi (grant no.
2019KY0522), the Open Research Fund from Guangxi Key Laboratory of
Liver Injury and Repair Molecular Medicine (grant no.
GXLIRMMKL-201802 and GXLIRMMKL-201816) and the Scientific Research
Project for Junior Faculty in Guilin Medical College (grant no.
2018glmcy055).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
GH designed and supervised the study. LD, YW, XH, CW
and AL conducted the experiments and collected the data. LD and GH
confirm the authenticity of all the raw data. LD and GH interpreted
the data and drafted the initial manuscript. All authors read and
approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Herbst RS, Morgensztern D and Boshoff C:
The biology and management of non-small cell lung cancer. Nature.
553:446–454. 2018.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Manning BD and Toker A: AKT/PKB signaling:
Navigating the network. Cell. 169:381–405. 2017.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Saxton RA and Sabatini DM: mTOR signaling
in growth, metabolism, and disease. Cell. 168:960–976.
2017.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Marenholz I, Zirra M, Fischer DF,
Backendorf C, Ziegler A and Mischke D: Identification of human
epidermal differentiation complex (EDC)-encoded genes by
subtractive hybridization of entire YACs to a gridded keratinocyte
cDNA library. Genome Res. 11:341–355. 2001.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Fagerberg L, Hallström BM, Oksvold P,
Kampf C, Djureinovic D, Odeberg J, Habuka M, Tahmasebpoor S,
Danielsson A, Edlund K, et al: Analysis of the human
tissue-specific expression by genome-wide integration of
transcriptomics and antibody-based proteomics. Mol Cell Proteomics.
13:397–406. 2014.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Jeng EE, Bhadkamkar V, Ibe NU, Gause H,
Jiang L, Chan J, Jian R, Jimenez-Morales D, Stevenson E, Krogan NJ,
et al: Systematic identification of host cell regulators of
legionella pneumophila pathogenesis using a genome-wide CRISPR
Screen. Cell Host Microbe. 26:551–563.e6. 2019.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Wei YJ, Hu QQ, Gu CY, Wang YP, Han ZG and
Cai B: Up-regulation of NICE-3 as a novel EDC gene could contribute
to human hepatocellular carcinoma. Asian Pac J Cancer Prev.
13:4363–4368. 2012.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Wang W, Li A, Han X, Wang Q, Guo J, Wu Y,
Wang C and Huang G: DEPDC1 up-regulates RAS expression to inhibit
autophagy in lung adenocarcinoma cells. J Cell Mol Med.
24:13303–13313. 2020.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Li A, Wang Q, He G, Jin J and Huang G: DEP
domain containing 1 suppresses apoptosis via inhibition of A20
expression, which activates the nuclear factor κB signaling pathway
in HepG2 cells. Oncol Lett. 16:949–955. 2018.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Chandrashekar DS, Bashel B, Balasubramanya
SAH, Creighton CJ, Ponce-Rodriguez I, Chakravarthi BVSK and
Varambally S: UALCAN: A portal for facilitating tumor subgroup gene
expression and survival analyses. Neoplasia. 19:649–658.
2017.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Okayama H, Kohno T, Ishii Y, Shimada Y,
Shiraishi K, Iwakawa R, Furuta K, Tsuta K, Shibata T, Yamamoto S,
et al: Identification of genes upregulated in ALK-positive and
EGFR/KRAS/ALK-Negative lung adenocarcinomas. Cancer Res.
72:100–111. 2012.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Győrffy B, Surowiak P, Budczies J and
Lánczky A: Online survival analysis software to assess the
prognostic value of biomarkers using transcriptomic data in
non-small-cell lung cancer. PLoS One. 8(e82241)2013.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Edge SB, Byrd DR, Compton CC, Fritz AG,
Greene FL and Trotti A (eds): AJCC Cancer Staging Manual. 7th
edition. Springer, New York, NY, 2010.
|
|
15
|
Wei Y, Zou Z, Becker N, Anderson M,
Sumpter R, Xiao G, Kinch L, Koduru P, Christudass CS, Veltri RW, et
al: EGFR-mediated beclin 1 phosphorylation in autophagy
suppression, tumor progression, and tumor chemoresistance. Cell.
154:1269–1284. 2013.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Levine B and Kroemer G: Biological
functions of autophagy genes: A disease perspective. Cell.
176:11–42. 2019.PubMed/NCBI View Article : Google Scholar
|
|
18
|
White E: The role for autophagy in cancer.
J Clin Invest. 125:42–46. 2015.PubMed/NCBI View
Article : Google Scholar
|
|
19
|
Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh
H, Troxel A, Rosen J, Eskelinen EL, Mizushima N, Ohsumi Y, et al:
Promotion of tumorigenesis by heterozygous disruption of the beclin
1 autophagy gene. J Clin Invest. 112:1809–1820. 2003.PubMed/NCBI View
Article : Google Scholar
|
|
20
|
Yue Z, Jin S, Yang C, Levine AJ and Heintz
N: Beclin 1, an autophagy gene essential for early embryonic
development, is a haploinsufficient tumor suppressor. Proc Natl
Acad Sci USA. 100:15077–15082. 2003.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Degenhardt K, Mathew R, Beaudoin B, Bray
K, Anderson D, Chen G, Mukherjee C, Shi Y, Gélinas C, Fan Y, et al:
Autophagy promotes tumor cell survival and restricts necrosis,
inflammation, and tumorigenesis. Cancer Cell. 10:51–64.
2006.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Chin YR and Toker A: Function of Akt/PKB
signaling to cell motility, invasion and the tumor stroma in
cancer. Cell Signal. 21:470–476. 2009.PubMed/NCBI View Article : Google Scholar
|