Introduction
Autosomal dominant intellectual disability (ID),
previously known as mental retardation, is a series of Mendelian
neurodevelopmental disorders that result in ID (1), and most of the cases are caused by
de novo heterozygous mutations in >50 genes [one gene per
case; Mendelian Inheritance in Man (MIM) phenotypic series no.
PS156200] and have a sporadic epidemiology. In general, patients
with a global development delay (GDD), which is defined as failure
to achieve developmental milestones within the expected age range
in at least two realms of development (2), have an increased risk of developing
ID, with an incidence of 1-3%, as they have altered development of
cognitive and adaptive functions (3). In addition, ID has been classified
into syndromic intellectual disability (S-ID) and non-S-ID (NS-ID),
and NS-ID has been traditionally defined by the presence of ID as
the sole clinical feature. Thus, the diagnosis of a dominant NS-ID
is much more difficult in clinical practice.
The Deciphering Developmental Disorders (DDD) Study
(4) first identified heterozygous
mutations in the chromosome alignment maintaining phosphoprotein 1
(CHAMP1) gene that caused a type of GDD/ID named autosomal dominant
mental retardation-40 (MRD40; MIM no. 616579), and the disorder was
previously known as NS-ID (Orphanet no. 178469) due to the lack of
more subtle neurological anomalies and psychiatric disorders shared
by the patients. As more MRD40 cases have been described (5), a better understanding of the disease
is on the horizon; however, genetic testing is now the only method
to diagnose MRD40.
The present study reported on a Chinese pediatric
patient diagnosed with MRD40 due to a CHAMP1 truncating mutation
identified by whole-exome sequencing (WES). A practical online
workflow of a comprehensive trio WES analysis was used in the
present study, as recommended by the DDD study (6).
Case study
The patient was a four-month-old male admitted to
the Third Affiliated Hospital of Zhengzhou University (Zhengzhou,
China) due to delayed development. His anomalies in appearance
included a tented upper lip, a high-arched palate and open-mouth
appearance (Fig. 1A),
hypertelorism, low-set ears (Fig.
1B) at the age of 4 months. At 9 months these included sparse
hair in general and microcephaly [occipitofrontal circumference
(OFC), 42.5 cm which was between -3 and -2 standard deviation (SD)
compared with a normal population] (7) (Fig.
1C). The neurological examination (8) revealed hypotonia, poor attention and
no active grasping consciousness. According to the Griffiths
Development Scales-Chinese (8), all
developmental parameters of the patient were equal to infants aged
1.5 months, indicating severe GDD.
The patient was the first-born child and delivered
by full-term, natural labor, and a family history of diseases in
the neurological system was not present.
Laboratory tests indicated slightly elevated urine
oxalic acid, succinic acid and glyceric acid levels; brain MRI
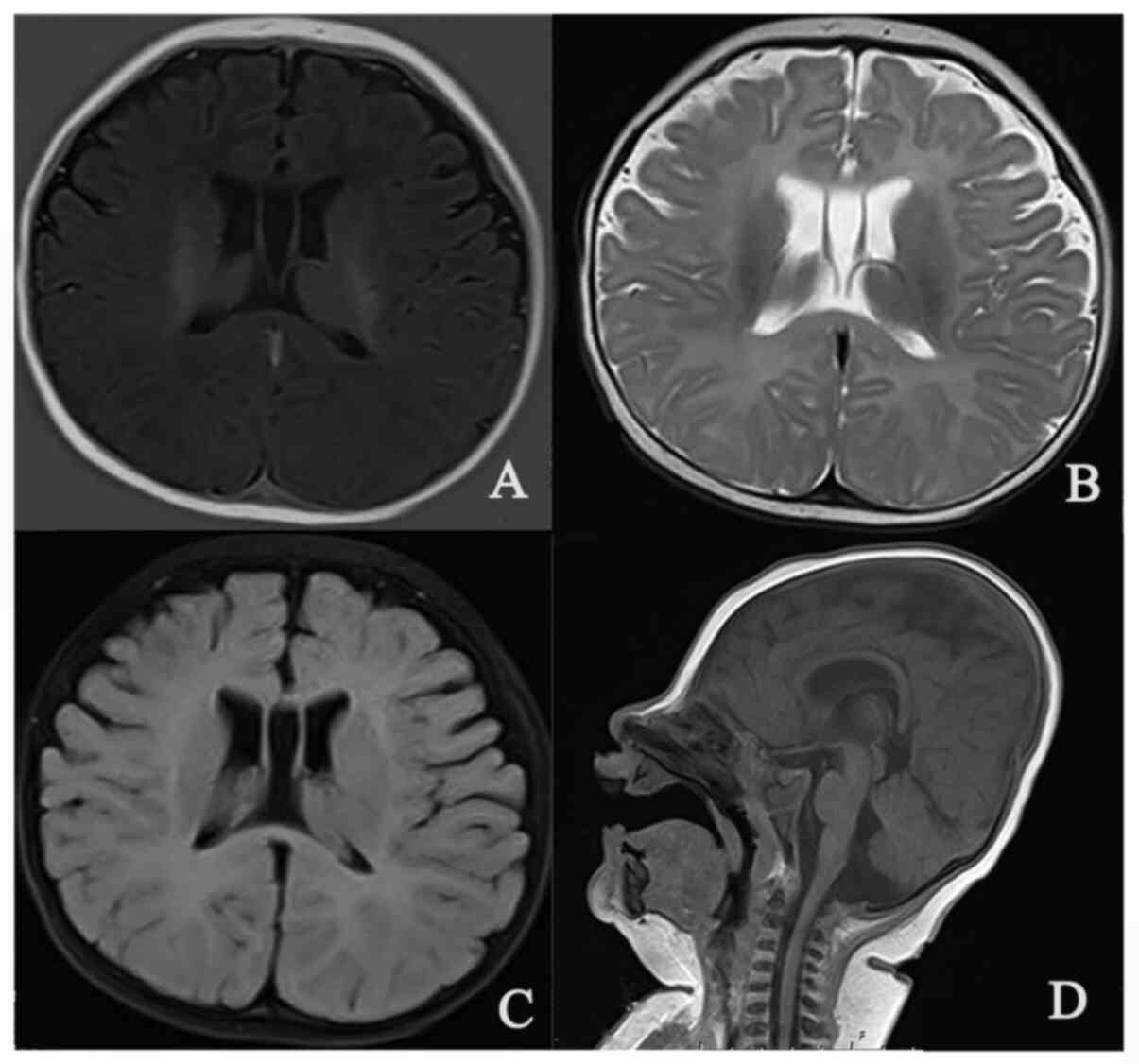
revealed symmetric enlargement in the bilateral lateral ventricle
(Fig. 2), and an 8-h video
electroencephalography test was unremarkable.
The patient was provided with proper rehabilitation
training, but at the age of 1 year and 3 months, the patient still
had difficulties acquiring language skills and was not able to
stand up by himself. In addition, the patient was monitored
continuously. The patient displayed a severe developmental delay at
the age of 9 months with microcephaly (Fig. 1C). The reexamination of organic acid
levels in urine provided normal results. At the time writing of the
present study, the patient was 1 year and 3 months old with an OFC
of 45 cm (-2 SD~-1 SD) and a body length of 67 cm (<-3 SD). The
average body length of a child of this age was 79.1±5 cm (7) but the weight of the patient was within
the normal range (11 kg). The patient was unable to communicate
with his family, in contrast to other children of the same age; he
still had difficulty acquiring language skills and was not able to
stand by himself.
Materials and methods
Methods
EDTA-treated peripheral blood samples for the
trio-WES analysis were collected from the patient and the patient's
parents to detect germline variation. Genomic DNA was extracted
from blood samples using the Blood Genome Column Medium Extraction
Kit (Kangweishiji). Protein-coding exome enrichment was performed
using xGen Exome Research Panel v1.0 (Integrated DNA Technologies),
which consists of 429,826 individually synthesized and
quality-controlled probes that target a 39-Mb protein-coding region
(19,396 genes) of the human genome and cover 51 Mb of end-to-end
tiled probe space. High-throughput sequencing was performed using
an Illumina NovaSeq 6000 series sequencer (PE150; Illumina, Inc.).
The sequencing process was performed by the Beijing Chigene
Translational Medicine Research Center Co., Ltd. Variants were
called and files in binary alignment map format were screened for
insertions/deletions and recalibrated using The Genome Analysis
Toolkit (9). Sequenced data were
aligned to the reference human genome (hg19; http://genome.ucsc.edu/index.html) and no control
sample was used.
After sequencing, the data were processed using the
Illumina DNA sequencing data analysis pipeline (https://www.illumina.com/informatics/sequencing-data-analysis/dna.html)
and sequencing data and phenotypes were normalized. Human Phenotype
Ontology terms were uploaded to the cloud platform for the genetic
disease analysis system (http://www.chigene.org). The data, including
pathogenicity of variants, inheritance type and phenotypes, were
determined using a protocol similar to that in the DDD study
(10) with the Chigene®
system, followed by manual screening by physicians.
Literature review
For the literature review, the PubMed (https://pubmed.ncbi.nlm.nih.gov/), Online MIM
(OMIM; https://omim.org/), Orphanet (https://www.orpha.net/consor/cgi-bin/index.php), The
Genetic and Rare Diseases Information Center (https://rarediseases.info.nih.gov/), Genetics
Home Reference (https://ghr.nlm.nih.gov/), GeneReviews (https://www.ncbi.nlm.nih.gov/books/NBK1116) and
Wanfang (Chinese, http://www.wanfangdata.com.cn/) databases were
searched using the key words ‘CHAMP1’ or ‘autosomal dominant mental
retardation’. Eligible studies were included if they fulfilled the
following criteria: A case report about autosomal dominant mental
retardation causing by CHAMP1 mutation. Exclusion criteria were
shown as follow: i) Publications not diagnosed as CHAMP1 mutation
associated autosomal dominant mental retardation; ii) studies
published in the format of meetings or as only an abstract.; or
iii) the cases had syndromic intellectual disability. All 17 cases
were included in the article ranging from the year 2015-2017
(Table I).
 | Table ISummary of patients with mutations in
chromosome alignment maintaining phosphoprotein 1. |
Table I
Summary of patients with mutations in
chromosome alignment maintaining phosphoprotein 1.
| Case no.a | Author (year) | Nationality of the
case | Age | Sex | GDD/ID | Feeding
difficulty | Seizures | Delay in motor
development | Speech delay | Abnormal
behavior | Muscular
hypotonia | Spasticity | Abnormal vision | Abnormal hearing | Dysmorphic
features | (Refs.) |
|---|
| 1 | Okamoto (2017) | Japanese | 6 y | M | + | + | + | + | + | + | - | + | + | - | + | (14) |
| 2 | Isidor (2016) #1 | French | NA | M | + | + | - | + | + | - | + | - | + | - | + | (13) |
| 3 | Isidor (2016) #2 | UK | NA | M | + | - | - | + | + | - | + | - | + | - | - | (13) |
| 4 | Isidor (2016) #3 | USA | NA | F | + | NA | + | + | + | + | + | - | + | - | + | (13) |
| 5 | Isidor (2016) #4 | USA | NA | M | | + | - | + | + | + | + | - | + | - | + | (13) |
| 6 | Isidor (2016) #5 | UK | NA | F | + | + | - | + | + | + | + | - | NA | - | + | (13) |
| 7 | Isidor (2016) #6 | UK | NA | F | + | NA | NA | + | + | - | + | - | + | - | + | (13) |
| 8 | Hempel (2015)
#1 | NA | 4 y | M | + | + | - | + | + | + | + | - | + | - | + | (6) |
| 9 | Hempel (2015)
#2 | Dutch | 3 y | M | + | + | + | + | + | + | + | - | + | - | + | (6) |
| 10 | Hempel (2015)
#3 | Dutch | 18 y | M | + | + | - | + | + | + | + | - | + | - | + | (6) |
| 11 | Hempel (2015)
#4 | Dutch | 3 y | F | + | + | - | + | + | + | + | - | + | - | + | (6) |
| 12 | Hempel (2015)
#5 | German | 9 y | F | + | - | - | + | + | + | + | - | + | - | + | (6) |
| 13 | Tanaka (2016)
#1 | NA | 23 y | F | + | + | - | + | + | + | + | + | - | + | + | (12) |
| 14 | Tanaka (2016)
#2 | NA | 7 y | F | + | + | + | + | + | + | - | + | + | - | + | (12) |
| 15 | Tanaka (2016)
#3 | NA | 4 y | F | + | + | - | + | + | + | + | + | + | + | + | (12) |
| 16 | Tanaka (2016)
#4 | NA | 12 y | F | + | + | + | + | + | + | + | - | + | - | + | (12) |
| 17 | Tanaka (2016)
#5 | NA | 6 y | F | + | + | - | + | + | + | + | - | + | + | + | (12) |
| 18 | Dong (2020) | Chinese | 4 m | M | + | - | - | + | + | - | + | - | - | - | + | Current |
Results
Molecular evaluation
A novel de novo mutation, NM_032436.2
(CHAMP1): c.530delCinsTTT p. Ser177Phefs*2, was identified in the
patient and it was confirmed using Sanger sequencing (Fig. 3). This CHAMP1 variation is a
loss-of-function mutation, and it is a de novo variation
verified by the patients parents who did not have this variation.
It has not been reported in the Single Nucleotide Polymorphism
Database (dbSNP; http://www.ncbi.nlm.nih.gov/SNP), the Exome
Aggregation Consortium (ExAC; http://exac.broadinstitute.org) or the 1,000 Genomes
Project (http://browser.1000genomes.org/index.html) database
(Minimum allele frequency <0.005), so it is a pathogenic
mutation according to the American College of Medial Genetics and
Genomics (ACMG) guidelines (PVS1+PS2+PM2) (11).
According to the results, the patient was diagnosed
with MRD40. Of note, the pediatric patient of the present study was
the first Chinese case to be reported to have a CHAMP1
mutation.
Literature review
At the time the present study was submitted, 18
cases of MRD40 had been reported, including the present case, and
this patient was the first known affected individual in China. As a
result, GDD/ID with speech delay, motor developmental delay and
facial anomalies were observed in all patients, as well as
hypotonia (17/18), abnormal muscular tone (16/18), vision damage
(15/18), abnormal behaviors (14/18) and reproductive issues (13/18)
were quite common in patients with MRD40, while seizures (4/18),
abnormal hearing (3/18) and spasticity (4/18) were less prevalent
phenotypes. The clinical features of the above-mentioned patients
are listed in Table I (6,12-14).
In addition, all identified mutations in the CHAMP1 gene in
patients diagnosed with MRD40 are truncating (nonsense or
frameshift) mutations (Table
II).
| Table IIMutations in CHAMP1 and brain MRI
features of patients with de novo CHAMP1 mutations. |
Table II
Mutations in CHAMP1 and brain MRI
features of patients with de novo CHAMP1 mutations.
| Case no. | DNA change | Protein change | Mutation types | Brain MRI
features |
|---|
| 1 |
c.2068_2069delGA |
p.Glu690Serfs*5 | Frameshift | Cavum septum
pellucidum, cavum vergae, cerebral atrophy, and decreased white
matter volume |
| 2 | c.1880C>G | p.Ser627* | Nonsense | Normal or
unremarkable findings |
| 3 | c.1002G>A | p.Trp334* | Nonsense | Normal or
unremarkable findings |
| 4 |
c.1876_1877delAG |
p.Ser626Leufs*4 | Frameshift | Normal or
unremarkable findings |
| 5 | c.1043G>A | p.Trp348* | Nonsense | Normal or
unremarkable findings |
| 6 | c.958_959delCC | p.Pro320* | Frameshift | Normal or
unremarkable findings |
| 7 | c.1489C>T | p.Arg497* | Nonsense | Normal or
unremarkable findings |
| 8 |
c.1866_1867delCA |
p.Asp622Glufs*8 | Frameshift | Mild brain atrophy
and cerebellar cortical dysplasia |
| 9 | c.1768C>T | p.Gln590* | Nonsense | Slightly delayed
myelination |
| 10 | c.1192C>T | p.Arg398* | Nonsense | Normal |
| 11 | c.635delC |
p.Pro212Leufs*7 | Frameshift | Normal |
| 12 | c.1192C>T | p.Arg398* | Nonsense | Normal |
| 13 | c.1044delG | p.Trp348* | Frameshift | Hypoplastic corpus
callosum |
| 14 | c.542_543delCT |
p.Ser181CysfsX5 | Frameshift | NA |
| 15 | c.1945C>T | p.Gln649* | Nonsense | Normal |
| 16 | c.1969C>T | p.Gln657* | Nonsense | Slightly decreased
white matter volume, possible hypopituitarism |
| 17 | c.2029G>T | p.Glu677* | Nonsense | Mild cerebellar
atrophy with mild inferior vermian hypogenesis |
| 18 |
c.530delCinsTTT |
p.Ser177Phefs*2 | Frameshift | Bilateral lateral
ventricle fullness |
Discussion
NS-ID refers to a rare, hereditary, neurological
disease characterized by early-onset cognitive impairment as the
sole disability (14); however,
patients with NS-ID may also be diagnosed with autism, epilepsy,
neuromuscular deficits or external deformity, which may explain why
MRD40 was previously considered an NS-ID (Orpha no. 178469). In
addition to GDD or ID, specific craniofacial dysmorphia, including
microcephaly, a long and hypotonic face, pointed chin, upslanting
palpebral fissures, short philtrum, high arched palate, tented
upper lip, everted lower lip and open mouth, facilitates the
diagnosis of MRD40(5). In addition,
previously reported epicanthal folds may be unspecific in Chinese
or East Asian patients, and eye dysfunction, such as strabismus and
hyperopia, are difficult to assess in infants (5). The abnormal behaviors mainly manifest
as hyperactivity that results in attention deficit hyperactivity
disorder or even self-injury; therefore, the abnormal behavior and
mental impairment of the patients should be taken seriously during
rehabilitation to care for patients. The patient of the present
study displayed dysmorphic features, severe motor and cognitive
developmental delays, as well as microcephaly. Most patients have
normal or nonspecific radiographs, while others exhibit slightly
delayed myelination, cerebral atrophy and a decreased white matter
volume (6,12-14).
For the patient of the present study, the bilateral lateral
ventricular enlargement appeared to be nonspecific, and no
abnormalities in the brain parenchyma were identified.
The human CHAMP1 protein is an 812-amino-acid
zinc-finger protein that is located on chromosome 13q34 and is
expressed in the fetal brain during development and in all adult
tissues (14). The protein contains
5 C-terminal C2H2 zinc-finger domains that were indicated to
regulate the binding of CHAMP1 to chromosomes on the mitotic
spindle in vitro, which has a key role in proper chromosome
alignment (10). All 18 individuals
with ID carrying CHAMP1 mutations reported to date had de
novo mutations, and 17 mutation sites were identified [two
patients carried the same mutation, namely c.1192C>T
(p.Arg398*)]. The CHAMP1 mutation identified in the patient of the
present study, c.530delCinsTTT (p.Ser177Phefs*2), has not been
reported previously, and it is a pathogenic mutation according to
ACMG guidelines (11). The reported
gene mutation types are nonsense and frameshift mutations, all of
which induced the synthesis of truncated proteins (Table II).
Thus, based on these findings, all of the truncating
mutations (Table II) would result
in loss of function of the CHAMP1 protein, which indicates the
requirement for the function of the lost C-terminal domains or
protein regions. Two regions, aa451-590 (FPE motifs, the region
localized to the spindle and kinetochores) and aa591-812
[containing zinc-finger (ZNF) domains], localize to the spindle and
both the chromosomes and the spindle, respectively (15), which are essential for proper
chromosome alignment. In addition to the disruption of
microtubule-kinetochore attachment, another functional study
indicated that truncated CHAMP1 proteins are unable to bind to two
of the direct binding partners of normal CHAMP1 protein, pogo
transposable element derived with ZNF domain and heterochromatin
protein 1, suggesting a pathogenic mechanism mediated by direct
interacting proteins of CHAMP1, several of which are involved in
syndromic ID (12). In summary,
functional loss of the C-terminus of CHAMP1 alters the localization
to chromosomes and spindles and disrupts the microtubule attachment
complex, which results in kinetochore-microtubule-related syndromic
ID.
WES is a powerful tool for the diagnosis of
neurodevelopmental disorders; however, the analysis of massive
amounts of data has been a major challenge (16). MRD40 was first described and the
phenotype was linked to CHAMP1 in the DDD study (4). The DDD project recommended a
comprehensive and highly efficient workflow for diagnosing
Mendelian diseases (6). The
workflow includes 3 main steps: i) Obtaining sequencing data from
the patient and the parents (trio) were preferred to determine the
inheritance of the candidate genetic variants; ii) assessment of
the pathogenicity of variants; and iii) matching the phenotype.
Using this approach, thousands of yield variant data may be
narrowed to 1-3 candidate genes. In the present study, the 3-step
screen was automatically performed using an online analysis system
(http://www.chigene.org), and in addition to
integrating genetic variations and disease databases which have
been anonymized to preserve confidentiality and references in the
pipeline, the system is online, which provides unlimited access
only for the treating physicians of the patients to participate in
the process and generate the final report. The limitation of the
present study was that sequenced data were aligned to hg19
(http://genome.ucsc.edu/index.html)
and no control sample was used. According to the Abstract, the
present study expands the spectrum of the known pathogenic
variants. The present case report and literature review also help
to improve the syndromic profile of the rare MRD40 disorder and
provides an example for the clinical diagnosis of MRD40. In
summary, a comprehensive, highly efficient workflow is key to
diagnosing rare genetic diseases.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request. The sequencing data have been uploaded to a curated
repository (https://www.ncbi.nlm.nih.gov/clinvar/submitters/507898).
Authors' contributions
YD made substantial contributions to conception and
design, agreed to be accountable for all aspects of the work in
ensuring that questions related to the accuracy or integrity of any
part of the work are appropriately investigated and resolved. XS
participated in drafting the manuscript acquired, analyzing and
interpreting the data. KD, TJ and JW participated in acquisition
and interpretation of data, revising the manuscript critically for
important intellectual content. RX, LW and RH participated in
analyzing and interpreting the data. YD and KD confirm the
authenticity of all the raw data. All authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
All procedures performed in studies involving human
participants were conducted in accordance with the ethical
standards of the institutional and/or national research committee
and with the 1964 Declaration of Helsinki and its later amendments
or comparable ethical standards. Ethical approval for the present
study was obtained from the Ethics Committee of the Third
Affiliated Hospital of Zhengzhou University (Zhengzhou, China; no.
2019116).
Patient consent for publication
Informed consent was obtained from the parents of
the patient included in the study for genetic testing and
publication of data and images.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Wieczorek D: Autosomal dominant
intellectual disability. Med Genet. 30:318–322. 2018.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Ferreira CR: The burden of rare diseases.
Am J Med Genet A. 179:885–892. 2019.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Ilyas M, Mir A, Efthymiou S and Houlden H:
The genetics of intellectual disability: Advancing technology and
gene editing. F1000Res. 9(F1000 Faculty Rev-22)2020.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Vasudevan P and Suri M: A clinical
approach to developmental delay and intellectual disability. Clin
Med (Lond). 17:558–561. 2017.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Deciphering Developmental Disorders Study.
Large-scale discovery of novel genetic causes of developmental
disorders. Nature. 519:223–228. 2015.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Hempel M, Cremer K, Ockeloen CW,
Lichtenbelt KD, Herkert JC, Denecke J, Haack TB, Zink AM, Becker J,
Wohlleber E, et al: De novo mutations in CHAMP1 cause intellectual
disability with severe speech impairment. Am J Hum Genet.
97:493–500. 2015.PubMed/NCBI View Article : Google Scholar
|
|
7
|
WHO Multicentre Growth Reference Study
Group. WHO Child Growth Standards based on length/height, weight
and age. Acta Paediatr Suppl. 450:76–85. 2006.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Tso WWY, Wong VCN, Xia X, Faragher B, Li
M, Xu X, Ao L, Zhang X, Jiao FY, Du K, et al: The Griffiths
development Scales-Chinese (GDS-C): A cross-cultural comparison of
developmental trajectories between Chinese and British children.
Child Care Health Dev. 44:378–383. 2018.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Van der Auwera GA, Carneiro MO, Hartl C,
Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen
D, Thibault J, et al: From FastQ data to high confidence variant
calls: The Genome Analysis Toolkit best practices pipeline. Curr
Protoc Bioinformatics. 43:11.10.1–11.10.33. 2013.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Wright CF, McRae JF, Clayton S, Gallone G,
Aitken S, FitzGerald TW, Jones P, Prigmore E, Rajan D, Lord J, et
al: Making new genetic diagnoses with old data: Iterative
reanalysis and reporting from genome-wide data in 1,133 families
with developmental disorders. Genet Med. 20:1216–1223.
2018.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American College
of Medical Genetics and Genomics and the Association for Molecular
Pathology. Genet Med. 17:405–424. 2015.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Tanaka AJ, Cho MT, Retterer K, Jones JR,
Nowak C, Douglas J, Jiang YH, McConkie-Rosell A, Schaefer GB,
Kaylor J, et al: De novo pathogenic variants in CHAMP1 are
associated with global developmental delay, intellectual
disability, and dysmorphic facial features. Cold Spring Harb Mol
Case Stud. 2(a000661)2016.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Isidor B, Küry S, Rosenfeld JA, Besnard T,
Schmitt S, Joss S, Davies SJ, Lebel RR, Henderson A, Schaaf CP, et
al: De novo truncating mutations in the kinetochore-microtubules
attachment Gene CHAMP1 cause syndromic intellectual disability. Hum
Mutat. 37:354–358. 2016.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Okamoto N, Tsuchiya Y, Kuki I, Yamamoto T,
Saitsu H, Kitagawa D and Matsumoto N: Disturbed chromosome
segregation and multipolar spindle formation in a patient with
CHAMP1 mutation. Mol Genet Genomic Med. 5:585–591. 2017.PubMed/NCBI View
Article : Google Scholar
|
|
15
|
Itoh G, Kanno S, Uchida KS, Chiba S,
Sugino S, Watanabe K, Mizuno K, Yasui A, Hirota T and Tanaka K:
CAMP (C13orf8, ZNF828) is a novel regulator of
kinetochore-microtubule attachment. EMBO J. 30:130–144.
2011.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Zhu B, Wu J, Chen G, Chen L and Yao Y:
Whole exome sequencing identifies a novel mutation of TPK1 in a
Chinese Family with Recurrent ataxia. J Mol Neurosci. 70:1237–1243.
2020.PubMed/NCBI View Article : Google Scholar
|