Introduction
Colon adenocarcinoma (COAD) is the most common
gastrointestinal malignancy in China and the third leading cause of
cancer-related death in the US, which affects >1 million
individuals annually worldwide (1).
It has been reported that COAD develops due to the progressive
accumulation of genetic and epigenetic alterations (2). COAD displays an aggressive behavior
and patients with COAD have poor survival. According to global
cancer statistics, new cases of COAD in 2018 accounted for 6.1% of
all newly diagnosed cancer cases and 5.8% of all cancer-associated
deaths (3). COAD is frequently
diagnosed at an advanced stage, as patients may be asymptomatic at
the early stage (3). COAD may be
treated with surgical techniques, chemotherapy and radiotherapy
(4). Although these treatments have
been improved in recent years, the prognosis for patients with COAD
with metastasis remains unsatisfactory (4). Therefore, it is necessary to expand
the current understanding of the disease and identify and apply
disease-specific biomarkers and therapeutic targets for COAD to
improve treatment outcomes.
Autophagy is an important and essential cellular
mechanism that has a key role in cellular degradation and the
recycling process in all eukaryotes. Numerous studies have
indicated that autophagy is induced under stress conditions to
degrade misfolded or aggregated proteins and clear damaged
organelles, leading to cell survival and cellular maintenance
(5). Autophagy is also involved in
numerous biological functions, including cellular differentiation,
development and cell defence. Thus, autophagy is primarily a
cytoprotective mechanism; however, excessive self-degradation may
be harmful (6). It has been
suggested that defects in autophagy regulation are associated with
several diseases, including cancer, neurodegeneration and metabolic
diseases (7). It has been reported
that autophagy may suppress or promote tumor growth depending on
the developmental stage and tumor type (8). Autophagy has an important role in
tumor suppression at early stages by protecting the cells against
inflammation, oxidative stress and DNA damage. However, autophagy
may also induce proliferation and metastasis of cancer cells.
Increasing evidence indicates that autophagy is increased during
chemotherapy, which leads to drug resistance and refractory cancer.
Thus, it is important to comprehensively analyze the expression of
autophagy-related genes (ARGs) in COAD.
The Cancer Genome Atlas (TCGA) is a web-based
database that was developed to discover and explore the major
cancer-causing genomic alterations to elucidate the mechanisms of
cancer development and progression (9). The Genotype-Tissue Expression (GTEx)
database was established to explore the correlation between human
genetic variations and tissue-specific gene expression in
non-diseased individuals (10). In
the present study, differentially expressed ARGs in COAD were
identified using the TCGA and GTEx databases. Enrichment analysis
and protein-protein interaction analysis of differentially
expressed ARGs were performed to improve the understanding of the
biological functions of these genes. Furthermore, the association
of the expression of ARGs with different clinicopathological
features was explored. Clinical and pathological data from our
hospital were used for further verification. From the individual
prognostic genes identified, a risk signature based on the
expression of these genes was developed.
Materials and methods
COAD datasets
Transcriptome data and clinical information were
downloaded from TCGA database, including 482 tumor samples and 42
normal samples. The GTEx database (https://www.gtexportal.org/home/index.html) contains
transcriptome data of various tissues from postmortem donors. The
transcriptome data of 308 normal colon samples were downloaded from
the GTEx database in December 2019.
Selection and download of ARGs
The Human Autophagy Database (HADb, http://www.autophagy.lu/) is a publicly available
project that provides structural and functional information on
autophagy-associated genes. All autophagy-associated genes listed
on this website are included in the present study, a list of which
(234 ARGs) was obtained from the HADb in December 2019.
Differentially expressed genes
(DEGs)
Transcriptome profiles of all COAD datasets were
merged and normalized. The R software was employed to search for
genes that were differentially expressed between different samples
[P<0.05, |log fold change (FC)| >1].
Construction and analysis of the
protein-protein interaction network
Protein-protein interactions (PPI) have a key role
in the majority of biological functions and processes. The Search
Tool for the Retrieval of Interacting Genes/proteins (STRING,
https://string-db.org/) is a web-based database
that is able to provide and predict the PPI networks. STRING was
used to construct the PPI network of the differentially expressed
ARGs (minimum required interaction score: Medium confidence=0.400).
Cytoscape software (https://cytoscape.org/), a free open-source platform
that serves as a tool for biological network analysis and
visualization, was used for PPI network analysis.
Enrichment analysis of differentially
expressed ARGs
The Kyoto Encyclopedia of Genes and Genomes (KEGG)
analysis is able to assign functional meanings to genes and genomes
at molecular and higher levels. Gene Ontology (GO) is a
comprehensive resource of computable knowledge regarding the
functions of genes and gene products. Metascape (http://metascape.org/gp/index.html) is a
web-based database that is able to provide a comprehensive gene
list annotation and analysis resources. Gene Set Enrichment
Analysis (GSEA; http://www.gsea-msigdb.org/gsea/index.jsp) is a
computational method that assesses gene expression data and
provides biological pathways. In the present study, enrichment
analysis of the differently expressed ARGs was performed using GSEA
and Metascape [miminum (Min) overlap=3, P<0.05 and Min
enrichment=1.5].
Prognostic value of differentially
expressed ARGs
Univariate Cox regression analysis was performed to
identify the differentially expressed ARGs that were significantly
associated with overall survival (OS). Multivariate Cox regression
analysis was used to search for genes that may be used as
independent prognostic indicators. Several candidate genes were
obtained for prognosis monitoring. The risk score for the signature
was calculated using the following formula: Risk score where
Coef i is the coefficient and Xi is the relative expression value
of each selected z-score-transformed gene expression value, divided
into high-risk and low-risk groups according to the median risk
score.
Immunohistochemical analysis
All samples were collected from The Third Hospital
of Hebei Medical University (Shijiazhuang, China). Clinical samples
were collected from December 2018 to September 2019 with written
informed consent from the patients. Immunohistochemical analysis
was performed using a tissue chip with a diameter of 4.0 mm
(Beijing Mairuibo Biotechnology Co., Ltd.). The array was heated in
sodium citrate buffer for 10 min in a microwave oven at 95˚C and
then sealed in normal goat serum (Beijing Mairuibo Biotechnology
Co., Ltd.) at 37˚C for 1 h. The samples were incubated with rabbit
anti-human autophagy-related 4B cysteine peptidase (ATG4B; 1:50
dilution; cat. no. A2981), death-associated protein kinase 1
(DAPK1; 1:50 dilution; cat. no. HPA048436) and Serpin family A
member 1 (Serpina1; 1:50 dilution; cat. no. SAB2109236; all from
Sigma-Aldrich; Merck KGaA) at 37˚C for 1 h. The samples were then
incubated with a goat anti-rabbit secondary antibody conjugated
with horseradish peroxidase (1:100 dilution; cat. no. F030212;
Beijing Biolab Technology Co., Ltd.) at 37˚C for 30 min. A total of
three pathologists, blinded to the patients' data, independently
analyzed the stained sections under a light microscope. The average
number of immune-positive cells in the specimen was determined
under a magnification of x400. The staining results were divided
into two categories as follows. a) Staining intensity: No staining,
0; buff, 1; dark yellow, 2; tan, 3; b) percentage of stained cells:
<1%, 0; 1-25%, 1; 25-50%, 2; 51-80%, 3; >80%, 4. The final
scores were added up and based on the staining scores, samples were
classified as low (final score 0-3, +), medium (final score 4-7,
++) or high (final score >7, +++). Image-Pro Plus software
(version 6; Media Cybernetics, Inc.) was employed for
immunohistochemical evaluation in the present study.
Statistical analysis
The χ2 test was used to compare the
distribution of clinicopathological parameters between the two risk
groups. The Mann-Whitney U test and one-way analysis of variance
followed by Bonferroni's post hoc test were used to compare the
risk scores of patients with different clinicopathological and
molecular pathological characteristics. Univariate and multivariate
Cox regression analyses were used to determine the prognostic value
of the risk score. To analyze the prediction efficiency, receiver
operating characteristic (ROC) curve analysis with the R package
‘survival ROC’ was employed. The OS of the patients was compared
using the Kaplan-Meier method with a two-sided log-rank test. R
software (version 3.5.3) and SPSS20.0 software (IBM Corp.) were
used to perform statistical analysis.
Results
Identification of differentially
expressed ARGs
The transcriptional profiles of 482 COAD samples and
350 non-tumor samples were downloaded from the TCGA and GTEx
databases and the expression of 234 ARGs was analyzed in these
samples (P<0.05, |logFC| >1). A total of 72 differentially
expressed ARGs were obtained (Fig.
1), comprising of 32 upregulated and 40 downregulated genes
(Fig. 1A and C). The heatmap of differentially expressed
ARGs was then drawn with R software (Fig. 1B).
PPI network
The PPI network was built on the basis of all DEGs
using the STRING online database and drawn with the software
Cytoscape (Fig. 1D). A total of 74
nodes and 509 edges were identified from PPI networks. The top 5
hub genes were CASP3, TP53, PIK3C3, GAPDH and BCL2L1.
Enrichment analysis of differentially
expressed ARGs
To understand the biological roles of the 72
differentially expressed ARGs, GO and KEGG analyses were performed
using Metascape and GSEA.
The GO terms significantly enriched in GSEA were
autophagosome, autophagosome membrane and cell body (Fig. 2A). KEGG analysis using GSEA revealed
that the significantly enriched pathways included pathways in
cancer, regulation of autophagy and apoptosis (Fig. 2B).
GO analysis using Metascape revealed that the
differentially expressed ARGs were mainly enriched in autophagy,
regulation of autophagy, organelle disassembly, ubiquitin-like
protein ligase binding, response to starvation, positive regulation
of programmed cell death, response to topologically incorrect
protein, cytokine-mediated signaling pathway, phagophore assembly
site, protein kinase binding, inclusion body, regulation of
reactive oxygen species metabolic process, response to
interferon-gamma, regulation of protein kinase activity, lysosome,
peptidyl-serine phosphorylation, protein kinase activity, protein
folding, and protein localization to the membrane and perinuclear
region of the cytoplasm (Fig. 2C).
KEGG analysis using Metascape revealed that the DEGs were mainly
enriched in autophagy-animal, mitophagy-animal, protein processing
in endoplasmic reticulum, toxoplasmosis, pathways in cancer,
NOD-like receptor signaling pathway, measles, ErbB signaling
pathway, Chagas disease (American trypanosomiasis), mTOR signaling
pathway, estrogen signaling pathway, Parkinson's disease,
endocytosis, Jak/STAT signaling pathway, Alzheimer's disease, HIF-1
signaling pathway, phagosome, Hippo signaling pathway and
regulation of actin cytoskeleton (Fig.
2D).
Identification of prognostic genes
The differently expressed ARGs were analyzed by to
identify the prognostic genes (Figs.
3 and 4). The analysis revealed
five prognostic genes (Fig. 3A). In
order to identify the genes with an independent prognostic
capability, multivariate Cox regression analysis was performed with
SPSS software (Fig. 3A). The ARGs
with a significant independent prognostic value were SERPINA1,
DAPK1 and ATG4B (Table I). DAPK1
and ATG4B were closely associated with low OS of patients with
COAD. Furthermore, downregulation of SERPINA1 was associated with
low OS in patients with COAD (Fig.
3C). Based on these genes, a risk score was calculated.
According to the median risk score, the COAD patients were divided
into a high-risk group and low-risk group. Kaplan-Meier plots were
employed to determine the performance of the risk score in
predicting the clinical outcome of patients with COAD. The results
suggested that the survival rate of the high-risk group was
significantly lower than that of the low-risk group (Fig. 3B). Furthermore, after adjusting for
clinicopathological features (age, gender, TNM, T, N and M stage)
by univariate and multivariate analyses, the risk score remained a
useful independent prognostic indicator (Fig. 4C and D). The risk score distribution of COAD
patients, the number of patients in different risk groups, a
thermogram of three prognostic genes and risk score distributions
in patients are provided in Fig.
3D. The prognostic value of the risk score in patients with
COAD was analyzed by ROC curve analysis, revealing an area under
the ROC curve of 0.646, with a cut-off value of 1.565 (Fig. 4B).
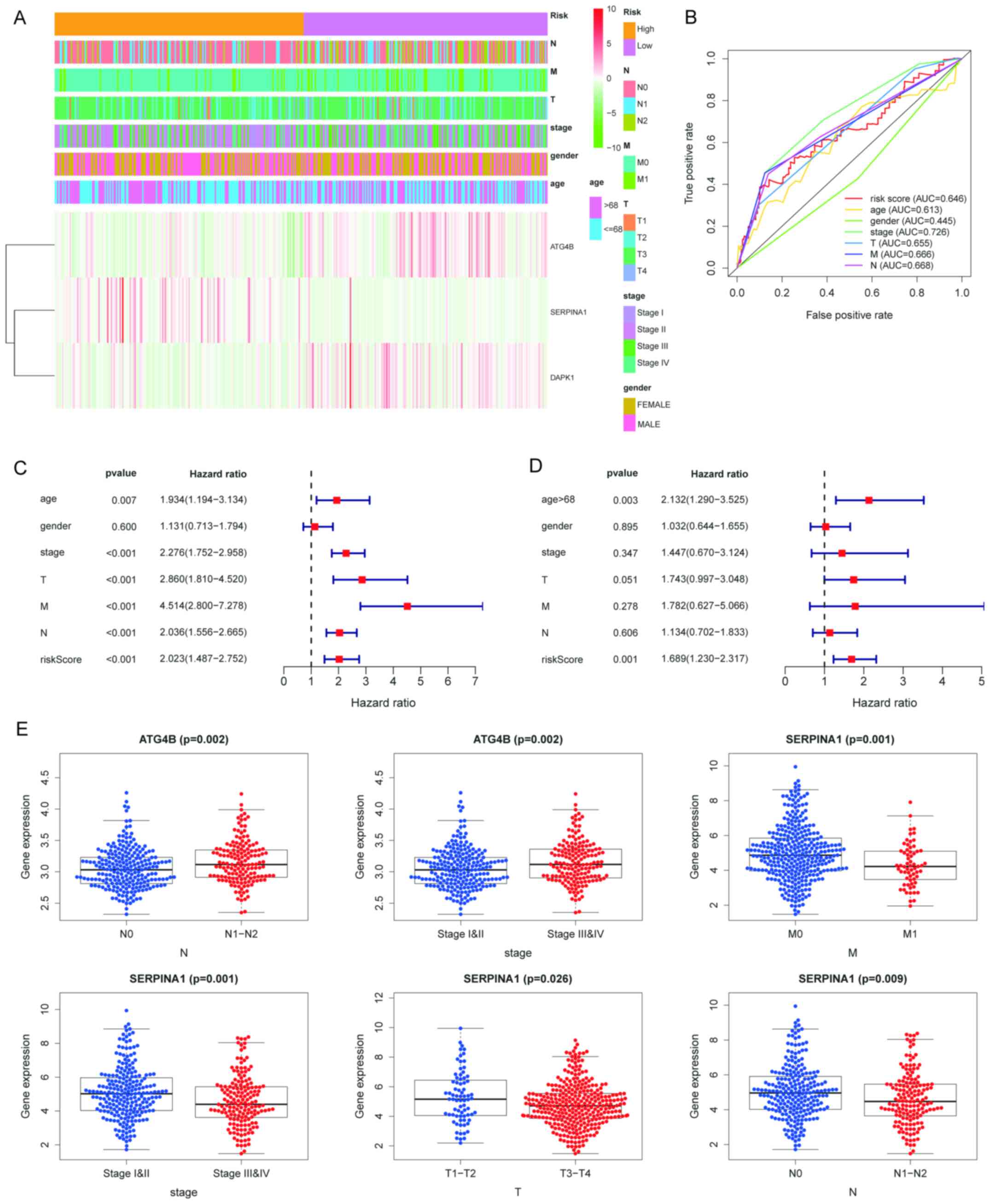 | Figure 4Verification of the accuracy of the
prognostic index model for ARGs. (A) Heatmap of colon cancer
datasets. (B) Multi-ROC curve from clinical trials. The cut-off
value for the age was 68 years. For stage, the difference between
stage I-Ⅱ and stage III-IV was compared. For the risk score, the
cut-off value was 1.565. (C) Univariate and (D) multivariate
analysis of the single parameters. Hazard ratios are provided with
95% CI. (E) Clinical relevance analysis of ATG4B and SERPINA1, the
expression value of genes was shown in boxplots. AUC, area under
the ROC curve; ROC, receiver operating characteristic; ARG,
autophagy-related gene; M, metastasis stage; N, nodal stage; T,
tumor stage; SERPINA1, Serpin family A member 1; ATG4B,
autophagy-related 4B cysteine peptidase. |
 | Table IGO summary for prognostic genes. |
Table I
GO summary for prognostic genes.
| Gene symbol | GO summary |
|---|
| SERPINA1 | GO:0048208 COPII
vesicle coating; GO:0048207 vesicle targeting, rough ER to
cis-Golgi; GO:0048199 vesicle targeting, to, from or within
Golgi |
| DAPK1 | GO:0071447 cellular
response to hydroperoxide; GO:2000310 regulation of NMDA receptor
activity; GO:0043280 positive regulation of cysteine-type
endopeptidase activity involved in apoptotic process |
| ATG4B | GO:0051697 protein
delipidation; GO:0000045 autophagosome assembly; GO:1905037
autophagosome organization |
The Mann-Whitney U test was performed to explore the
association between the risk score and clinicopathological
parameters. Regarding the metastasis (M) stage, the results
suggested that the risk score of the M1 group was higher than that
of the M0 group (P=0.009). Furthermore, the risk score in the TNM
stage III/IV group was higher than that in the stage I/II group
(P=0.001). Regarding the nodal (N) stage, the risk score in the
N1/N2 group was higher than that in the N0 group (P=0.001; Fig. 3E).
A heatmap depicting the expression of the three ARGs
in high-risk and low-risk patients in the TCGA dataset is presented
in Fig. 4A. The Mann-Whitney U test
indicated that high expression of ATG4B was significantly
associated with N (P=0.002) and advanced stages (P=0.002; Fig. 4E). Furthermore, a significant
association between low expression of SERPINA1 and advanced M stage
(P=0.001), advanced pathological stage (P=0.001), advanced
pathological T stage (P=0.026) and advanced N stage (P=0.009) was
determined (Fig. 4E).
Immunohistochemical analysis
Representative images of ATG4B, DAPK1 and SERPINA1
staining of COAD tissues are presented in Fig. 5. The clinicopathological data of the
patients are presented in Table SI
and the association of the expression levels with the patients'
clinicopathological characteristics are presented in Table II. A total of 75.6% (34/45)
patients were >60 years old, while 24.4% (11/45) were ≤60 years
old (age range, 34-88 years; median, 69 years). A total of 62.2%
(28/45) were males, while 37.8% (17/45) were females. The results
suggested that in COAD tissues, ATG4B expression was low in 20.0%
(9/45), moderate in 51.1% (23/45) and high in 28.9% (13/45) of
cases and DAPK1 expression was low in 33.3% (15/45), moderate in
64.4% (29/45) and high in 2.2% (1/45) of cases, whereas SERPINA1
expression was low in 2.2% (1/45), moderate in 84.4% (38/45) and
high in 13.3% (6/45) of cases. ATG4B expression was significantly
associated with age (P=0.0043) and SERPINA1 expression was
significantly associated with tumor size (P=0.0034; Table II).
| Table IIClinicopathological variables and the
expression of ATG4B, DAPK1 and SERPINA1. |
Table II
Clinicopathological variables and the
expression of ATG4B, DAPK1 and SERPINA1.
| | ATG4B (%) | DAPK1 (%) | SERPINA1 (%) |
|---|
| Parameters | n | -/+ | ++ | +++ | P-value | -/+ | ++ | +++ | P-value | -/+ | ++ | +++ | P-value |
|---|
| Age (years) | | | | | 0.0043 | | | | 0.7265 | | | | 0.0779 |
|
>60 | 34 | 3 | 20 | 11 | | 12 | 21 | 1 | | 0 | 28 | 6 | |
|
≤60 | 11 | 6 | 3 | 2 | | 3 | 8 | 0 | | 1 | 10 | 0 | |
| Sex | | | | | 0.1244 | | | | 0.6398 | | | | 0.6057 |
|
Female | 17 | 1 | 9 | 7 | | 5 | 12 | 0 | | 0 | 14 | 3 | |
|
Male | 28 | 8 | 14 | 6 | | 10 | 17 | 1 | | 1 | 24 | 3 | |
| Tumor size
(cm) | | | | | 0.7515 | | | | 0.0825 | | | | 0.0034 |
|
>5 | 37 | 8 | 18 | 11 | | 12 | 25 | 0 | | 1 | 34 | 2 | |
|
≤5 | 8 | 1 | 5 | 2 | | 3 | 4 | 1 | | 0 | 4 | 4 | |
| Lymphatic
metastasis | | | | | 0.1014 | | | | 0.5365 | | | | 0.2428 |
|
Yes | 17 | 2 | 7 | 8 | | 7 | 10 | 0 | | 1 | 15 | 1 | |
|
No | 28 | 7 | 16 | 5 | | 8 | 19 | 1 | | 0 | 23 | 5 | |
| Recurrence and
metastasis | | | | | 0.1217 | | | | 0.8593 | | | | 0.4660 |
|
Yes | 7 | 0 | 6 | 1 | | 2 | 5 | 0 | | 0 | 7 | 0 | |
|
No | 38 | 9 | 17 | 12 | | 13 | 24 | 1 | | 1 | 31 | 6 | |
Discussion
COAD is the most common subtype of colon cancer,
with high mortality and a small number of systemic treatment
options. Despite commendable advances in the treatment modalities
for COAD, it remains one of the most common causes of
cancer-associated death worldwide. Thus, it is essential to develop
novel and non-invasive diagnostic and prognostic biomarkers for
COAD at a molecular level (11-13).
Autophagy is a highly conserved pathway that has a key role in
cellular self-digestion to provide energy and metabolic precursors
under conditions of starvation and to maintain cellular homeostasis
(14). It has been reported that
autophagy is increased during tumorigenesis and progression of
various cancer types, including non-small cell lung cancer
(15), liver cancer (16) and breast cancer (17). Chen et al (18) indicated that autophagy has an
important role in the innate immune response and regulation of
autophagy in tumor-associated macrophages may limit cancer growth
and progression. A growing body of evidence indicates that
autophagy has a key role in multidrug resistance after long-term
chemotherapy, which may result in refractory cancer and tumor
recurrence (19-21).
In addition, autophagy promotes tumorigenesis and the development
of cancer cells through various mechanisms (22). Thus, modification of differentially
expressed ARGs may improve the responsiveness of cancer cells to
treatments and provide novel targeted therapy options for COAD. In
the present study, key prognostic ARGs in patients with COAD were
identified, which may be utilized for the treatment of COAD.
In recent years, with the continuous improvements in
next-generation sequencing technology and cost reduction,
bioinformatics analysis has been widely used for studying clinical
markers and identifying potential targets of diseases, including
diabetes and tumors (23).
Furthermore, the number of DNA and RNA sequences submitted to
public databases, including TCGA and gene expression omnibus (GEO),
has markedly increased in recent years. In the present study, 72
differentially expressed ARGs between COAD samples and normal
samples were identified. GO and KEGG analysis were performed using
Metascape and GSEA was applied to identify connections between
genes and the potential molecular mechanisms of COAD. Of note, it
was observed that the common term enriched in the KEGG analysis
using Metascape and GSEA was ‘Pathways in cancer’. The results
suggested that certain ARGs were closely associated with
tumorigenesis. It has been reported that autophagy is able to
inhibit cancer initiation at early stages, which may be due to
autophagy limiting oncogenic signaling and preventing the toxic
accumulation of organelles and damaged proteins (19,24).
However, Guo and White (25)
indicated that autophagy is induced in certain types of cancer and
cancer cells rely on autophagy to survive. Cancer cells may promote
autophagy-mediated recycling to maintain mitochondrial function and
energy homeostasis, which has a key role in regulating tumor growth
and proliferation (25). Therefore,
autophagy has complex and variable effects on tumor cells and has
different roles in different tumor types.
In the present study, three key prognostic ARGs
(SERPINA1, DAPK1 and ATG4B) were identified by univariate and
multivariate survival analyses. SERPINA1 encodes a serine protease
inhibitor whose targets include elastase, plasmin, thrombin,
trypsin, chymotrypsin and plasminogen activator. Specific mutations
in the SERPINA1 gene may lead to alpha-1-antitrypsin deficiency.
The Z variant of alpha-1-antitrypsin cannot be polymerized in the
endoplasmic reticulum of hepatocytes and cannot be secreted, which
may lead to hepatocellular carcinoma (26,27).
Furthermore, Griffith et al (28) indicated that SERPINA1 may be a
potential biomarker with sufficient sensitivity and specificity for
the diagnosis of thyroid tumors. However, the functional mechanism
of SERPINA1 in COAD is not clear. DAPK1 encodes a structurally
unique 160-kDa calmodulin-dependent serine-threonine kinase that
carries eight ankyrin repeats and two putative P-loop consensus
sites. DAPK1 is a positive regulator of gamma-interferon-induced
programmed cell death. Previous studies have indicated that DAPK1
is a candidate tumor suppressor and DAPK1methylation is a potential
biomarker for the early diagnosis of gastrointestinal cancer
(29). Furthermore, Singh et
al (30) reported that DAPK1
mediates a wide range of cellular processes such as apoptosis and
autophagy. However, the roles of DAPK1 in COAD have remained
largely elusive. ATG4B, a member of the autophagin protein family,
has a key role in cell homeostasis and cellular remodeling during
differentiation. Numerous studies have reported that targeting
ATG4B may suppress tumor growth by activating the AMP-activated
protein kinase energy-sensing pathway (31). Fu et al (32) indicated that ATG4B is an independent
positive regulator of tumor proliferation. These studies suggested
that ATG4B is a potential target for COAD.
With the development of large-scale public
databases, the identification of prognostic factors in cancer
patients based on expression spectrum analysis has been proposed.
For instance, Li et al (33)
identified 20 genes related to malignant tumors, such as
triple-negative breast cancer, COAD, ovarian cancer and
glioblastoma multiforme from the GEO database. Wan et al
(34) comprehensively analyzed 311
CRC samples from TCGA and GEO databases. However, these studies are
not combined with the corresponding clinical information and the
molecular markers obtained are of low prognostic value. In the
present study, transcriptome information was combined with
corresponding clinical information to obtain molecular markers with
prognostic value.
In conclusion, three key prognostic ARGs (SERPINA1,
DAPK1 and ATG4B) were identified by re-analyzing public datasets.
These genes may be potential biomarkers for COAD. In addition, a
novel risk score model was constructed based on the expression
levels and HR value of these genes, which may predict the survival
rate of patients with COAD.
Supplementary Material
Clinicopathological data of the
patients.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
RNA-seq data were downloaded for the TCGA COAD
cohort and the mRNA splicing pattern data were obtained with the
SpliceSeq tool from TCGA. The raw data are available from TCGA
(https://tcga-data.nci.nih.gov/tcga/)
and GTEx (https://www.gtexportal.org/home/index.html). In
addition, the data of the present cohort are available from the
corresponding author on reasonable request.
Authors' contributions
Conception and design: XZ, RX and JM. Administrative
support: WF. Collection and collation of data: JX. Data analysis
and interpretation: XZ, RX, JM, WF, YL and JX. Manuscript writing:
JM. Confirmation of the authenticity of the raw data: JM and XZ.
All authors read and approved the final version of the manuscript.
The authors are accountable for all aspects of the work in ensuring
that questions related to the accuracy or integrity of any part of
the work are appropriately investigated and resolved.
Ethics approval and consent to
participate
This study complied with the Declaration of Helsinki
and was approved by the Ethics Committees of The Third Hospital of
Hebei Medical University (Shijiazhuang, China). Written informed
consent was obtained from all patients.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Kuipers EJ, Grady WM, Lieberman D,
Seufferlein T, Sung JJ, Boelens PG, van de Velde CJ and Watanabe T:
Colorectal cancer. Nat Rev Dis Primers. 1(15065)2015.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Erickson LA: Adenocarcinoma of the colon
and microsatellite instability. Mayo Clin Proc. 93:669–670.
2018.PubMed/NCBI View Article : Google Scholar
|
|
3
|
White A, Joseph D, Rim SH, Johnson CJ,
Coleman MP and Allemani C: Colon cancer survival in the United
States by race and stage (2001-2009): Findings from the CONCORD-2
study. Cancer. 123 (Suppl 24):S5014–S5036. 2017.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Kriegsmann M, Longuespée R, Wandernoth P,
Mohanu C, Lisenko K, Weichert W, Warth A, Dienemann H, De Pauw E,
Katzenberger T, et al: Typing of colon and lung adenocarcinoma by
high throughput imaging mass spectrometry. Biochim Biophys Acta
Proteins Proteom. 1865:858–864. 2017.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Li X, He S and Ma B: Autophagy and
autophagy-related proteins in cancer. Mol Cancer.
19(12)2020.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Gomes LR, Menck C and Leandro GS:
Autophagy roles in the modulation of DNA repair pathways. Int J Mol
Sci. 18(2351)2017.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Su Z, Yang Z, Xu Y, Chen Y and Yu Q:
Apoptosis, autophagy, necroptosis, and cancer metastasis. Mol
Cancer. 14(48)2015.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Ravanan P, Srikumar IF and Talwar P:
Autophagy: The spotlight for cellular stress responses. Life Sci.
188:53–67. 2017.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Wang Z, Jensen MA and Zenklusen JC: A
Practical Guide to The Cancer Genome Atlas (TCGA). Methods Mol
Biol. 1418:111–141. 2016.PubMed/NCBI View Article : Google Scholar
|
|
10
|
GTEx Consortium: The Genotype-Tissue
Expression (GTEx) project. Nat Genet. 45:580–585. 2013.PubMed/NCBI View
Article : Google Scholar
|
|
11
|
Dienstmann R, Salazar R and Tabernero J:
Personalizing colon cancer adjuvant therapy: Selecting optimal
treatments for individual patients. J Clin Oncol. 33:1787–1796.
2015.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Rosen AW, Degett TH and Gögenur I:
Individualized treatment of colon cancer. UgeskrLaeger.
178(V11150916)2016.PubMed/NCBI(In Danish).
|
|
13
|
Colon Cancer. Am Fam Physician.
97(Online)2018.PubMed/NCBI
|
|
14
|
Fulda S and Kogel D: Cell death by
autophagy: Emerging molecular mechanisms and implications for
cancer therapy. Oncogene. 34:5105–5113. 2015.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Liu G, Pei F, Yang F, Li L, Amin AD, Liu
S, Buchan JR and Cho WC: Role of autophagy and apoptosis in
non-small-cell lung cancer. Int J Mol Sci. 18(367)2017.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Akkoc Y and Gozuacik D: Autophagy and
liver cancer. Turk J Gastroenterol. 29:270–282. 2018.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Han Y, Fan S, Qin T, Yang J, Sun Y, Lu Y,
Mao J and Li L: Role of autophagy in breast cancer and breast
cancer stem cells (Review). Int J Oncol. 52:1057–1070.
2018.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Chen P, Cescon M and Bonaldo P:
Autophagy-mediated regulation of macrophages and its applications
for cancer. Autophagy. 10:192–200. 2014.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Li YJ, Lei YH, Yao N, Wang CR, Hu N, Ye
WC, Zhang DM and Chen ZS: Autophagy and multidrug resistance in
cancer. Chin J Cancer. 36(52)2017.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Auberger P and Puissant A: Autophagy, a
key mechanism of oncogenesis and resistance in leukemia. Blood.
129:547–552. 2017.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Chen C, Lu L, Yan S, Yi H, Yao H, Wu D, He
G, Tao X and Deng X: Autophagy and doxorubicin resistance in
cancer. Anticancer Drugs. 29:1–9. 2018.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Bhat P, Kriel J, Shubha Priya B, Basappa
Shivananju NS and Loos B: Modulating autophagy in cancer therapy:
Advancements and challenges for cancer cell death sensitization.
Biochem Pharmacol. 147:170–182. 2018.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Chan LL and Jiang P: Bioinformatics
analysis of circulating cell-free DNA sequencing data. ClinBiochem.
48:962–975. 2015.PubMed/NCBI View Article : Google Scholar
|
|
24
|
White E, Mehnert JM and Chan CS:
Autophagy, metabolism, and cancer. Clin Cancer Res. 21:5037–5046.
2015.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Guo JY and White E: Autophagy, metabolism,
and cancer. Cold Spring Harb Symp Quant Biol. 81:73–78.
2016.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Lachaux A and Dumortier J: Hepatic
involvement in hereditary alpha-1-antitrypsin deficiency). Rev Mal
Respir. 31:357–364. 2014.PubMed/NCBI View Article : Google Scholar : (In French).
|
|
27
|
Mitchell EL and Khan Z: Liver disease in
alpha-1 antitrypsin deficiency: Current approaches and future
directions. Curr Pathobiol Rep. 5:243–252. 2017.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Griffith OL, Melck A, Jones SJ and Wiseman
SM: Meta-analysis and meta-review of thyroid cancer gene expression
profiling studies identifies important diagnostic biomarkers. J
Clin Oncol. 24:5043–5051. 2006.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Yuan W, Chen J, Shu Y, Liu S, Wu L, Ji J,
Liu Z, Tang Q, Zhou Z, Cheng Y, et al: Correlation of DAPK1
methylation and the risk of gastrointestinal cancer: A systematic
review and meta-analysis. PLoS One. 12(e0184959)2017.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Singh P, Ravanan P and Talwar P: Death
Associated Protein Kinase 1 (DAPK1): A regulator of apoptosis and
autophagy. Front Mol Neurosci. 9(46)2016.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Liu PF, Hsu CJ, Tsai WL, Cheng JS, Chen
JJ, Huang IF, Tseng HH, Chang HW and Shu CW: Ablation of ATG4B
suppressed autophagy and activated AMPK for cell cycle arrest in
cancer cells. Cell Physiol Biochem. 44:728–740. 2017.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Fu Y, Hong L, Xu J, Zhong G, Gu Q, Gu Q,
Guan Y, Zheng X, Dai Q, Luo X, et al: Discovery of a small molecule
targeting autophagy via ATG4B inhibition and cell death of
colorectal cancer cells in vitro and in vivo. Autophagy.
15:295–311. 2019.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Li M, Wang P, Zhang N, Guo L and Feng YM:
Identification of genes of four malignant tumors and a novel
prediction model development based on PPI data and support vector
machines. Cancer Gene Ther. 27:715–725. 2020.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Wan L, Yu W, Shen E, Sun W, Liu Y, Kong J,
Wu Y, Han F, Zhang L, Yu T, et al: SRSF6-regulated alternative
splicing that promotes tumour progression offers a therapy target
for colorectal cancer. Gut. 68:118–129. 2019.PubMed/NCBI View Article : Google Scholar
|


















