Introduction
Dysbiosis of the gut microbiota has an important
role in various diseases of the host, including obesity, diabetes,
inflammatory bowel disease, autoimmune diseases, acute severe
pancreatitis, hepatitis, liver cirrhosis, tumors and numerous other
pathological states (1-14).
A large number of studies focus on gut microbiota changes in liver
cirrhosis and acute liver injury [induced by carbon tetrachloride
(CCl4)] (1-6).
With the increasing understanding of the relationship between the
gut microbiota and host metabolism in cirrhosis, novel methods and
approaches may be developed to promote the treatment and prevention
of liver cirrhosis (1,2). A study indicated that the abundance of
Firmicutes increased and that of Bacteroidetes decreased over time
during CCl4-induced acute liver injury and that initial
alterations in the Firmicute/Bacteroidete ratio may be beneficial
to liver repair (6). However, the
changes of the intestinal microbiota during partial hepatectomy
(PH)-induced acute liver injury have remained elusive. Although 2/3
PH (~70%) is the most studied model of acute liver injury and liver
regeneration, resection of ~50% volume of the liver is more
frequent in the treatment of liver-associated tumors, as well as
living donor liver transplantation and split-liver transplantation
in the clinic (15,16). Previous studies suggested that 50%
PH caused an immediate and sustained increase in the allospecific
cytolytic response in C3H/HeJ mice (17) and caused obvious liver cell
acidophilic necrosis and the majority of hepatocyte nuclei to
increase in size with vesicular bodies, caryocinesia of nucleoli
and stromal inflammatory cell infiltration within 3 days
post-operation in C57/BL6 mice (18). In the present study, 50% PH was
performed in C57/BL6 mice to mimic the clinical conditions where
about half of the liver is surgically resected, and the changes of
the gut microbiota and associated pathways during acute liver
injury and repair following PH were analyzed. The present study may
lay a foundation for further studies on the gut-micbiotaliver
metabolic network and aid in establishing the role of probiotics in
hepatectomy.
Materials and methods
Animals
A total of 50 male wild-type C57/BL6 mice (body
weight, 20-25 g; age, 8 weeks) were purchased from Shanghai SLAC
Laboratory Animal Co., Ltd. Mice were raised and maintained in a
standardized environment (temperature, 21˚C; humidity, 60%) with a
12-h light/dark cycle and provided with food and water ad
libitum. The Animal Experimental Ethics Committee of the First
Affiliated Hospital, School of Medicine, Zhejiang University
(Hangzhou, China) approved all of the procedures. The mice were
divided into the following groups: No surgery control group [normal
control (NC) group; n=10], sham-operation group (Sham group; n=20)
and liver resection (LR) group (50% PH; n=20). All of the mice were
kept under the same conditions.
50% PH-induced acute liver injury
The mice were anesthetized by isoflurane inhalation
(anesthetic induction on 5% isoflurane at a flow of 10 l/min,
followed by anesthetic maintenance using an inspiratory fraction of
1.8-2.2% at a flow of 1 l/min) (19). The caudate lobe, left lateral lobe
and left median lobe were then resected to generate the LR model
(50% PH) (18,19).
Sample collection
After isoflurane inhalation anesthesia, blood
samples were collected by inferior vena cava puncture at 3 and 14
days post-operation. Specimens of the NC group were obtained at the
same time as those of Sham3 group. Following exsanguination, the
livers were harvested, fixed with 4% paraformaldehyde and
paraffin-embedded for histological analysis. The small intestinal
contents were precisely dissected and harvested, frozen in liquid
nitrogen and preserved at -80˚C until analysis. Serum was acquired
by centrifugation of the blood samples at 2,500 x g for 20 min at
4˚C and kept at -80˚C until analysis.
Biochemical analysis and liver
histology
Serum alanine aminotransferase (ALT) and aspartate
aminotransferase (AST) were determined using an automatic chemistry
analyzer (Mindray BS-220; Mindray Bio-medical Electronics Co.,
Ltd.) according to the manufacturer's protocol. For
histopathological analysis, paraffin-embedded liver samples were
sectioned (5 µm thickness) and stained with H&E. Images were
acquired (original magnification, x400) using a digital colour
camera (DP20; Olympus Corporation).
Intestinal microbiota analysis by 16S
ribosomal (r)RNA gene sequencing
The small intestinal contents from all the mice were
subjected to analysis of the microbial diversity. DNA was extracted
from all samples using an E.Z.N.A.® Stool DNA Kit (cat.
no. D4015-02; Omega Biotek). The V3-V4 region of the 16S rRNA gene
was amplified with primers as follows: 341 Forward,
5'-CCTACGGGNGGCWGCAG-3'; and 805 Reverse,
5'-GACTACHVGGGTATCTAATCC-3'. PCR amplification was performed in a
reaction mixture with a total volume of 25 µl, including 25 ng of
purified DNA, 12.5 µl PCR Premix (Phusion Hot start flex 2X Master
Mix; cat. no. M0536L; New England Biolabs, Inc.), 2.5 µl of each
primer and PCR-grade water to adjust the volume. The PCR cycling
conditions consisted of an initial denaturation step at 98˚C for 30
sec, 32 cycles of denaturation at 98˚C for 10 sec, annealing at
52˚C for 30 sec and extension at 72˚C for 45 sec, and a final
extension step at 72˚C for 10 min (20). PCR products were purified using
AMPure XT beads (cat. no. A63880; Beckman Coulter Genomics),
quantified with a Qubit dsDNA HS Assay kit (cat. no. Q32854;
Invitrogen; Thermo Fisher Scientific, Inc.), pooled in equimolar
ratios and then sequenced with a 2x300 bp paired end protocol on a
MiSeq PE300 platform (Illumina, Inc.).
Data analysis
Raw sequencing data were merged using the Fast
Length Adjustment of SHort reads software v1.2.8 (http://ccb.jhu.edu/software/FLASH/) based on
overlapping bases. Quality filtering of the raw tags was performed
to obtain the high-quality clean tags according to fqtrim v0.9.4
(https://zenodo.org/record/20552#.YMF-26aS3IU).
Chimeric sequences were filtered using Vsearch software v2.3.4
(https://zenodo.org/record/198592#.YMGAHaaS3IV). The
processed sequences were assigned to the same operational taxonomic
units (OTUs) at 97% identity (21,22).
Representative sequences were selected for each OTU and annotated
against the Ribosomal Database Project (RDP) classifier (http://rdp.cme.msu.edu/) and National Center for
Biotechnology Information (NCBI)-16s database (https://www.ncbi.nlm.nih.gov/gene/27471). The
effective sequences were analyzed by Quantitative Insights Into
Microbial Ecology (QIIME) software v1.8.0 (23,24).
α-diversity, including the indexes of Chao1, Observed OTUs, Goods
coverage, Shannon and Simpson, was calculated by QIIME to analyze
the complexity of species diversity for a sample (23,24).
β-diversity, referring to the species diversity between different
samples, including principal component analysis (PCA), unweighted
UniFrac principal coordinate analysis (PCoA) and unweighted UniFrac
distance-based non-metric multidimensional scaling analysis (NMDS),
were calculated by QIIME (23,24).
Linear discriminant analysis (LDA) effect size (LEfSe) was applied
to estimate the characteristic types of bacteria of each group
(25). Metagenome prediction and
functional differences among groups were investigated by the
Phylogenetic Investigation of Communities by the Reconstruction of
Unobserved States (PICRUSt) software based on the Greengenes
database (26). The output files
were further analyzed by STatistical Analysis of Metagenomic
Profile (STAMP) software (27).
Statistical analysis
Values are expressed as the mean ± standard
deviation or the median (interquartile range) and variables were
compared between two groups by Student's t-test (normal
distribution and equal variance) or nonparametric Mann-Whitney
U-test (non-normal distribution). Comparisons among more than two
groups were performed by ANOVA with LSD post-hoc test or
nonparametric Kruskal-Wallis H-test. A two-tailed P<0.05 was
considered to indicate statistical significance. Data were analyzed
by SPSS 22.0 (IBM Corporation) and R software (v3.5.1; 2018; R
Development Core Team).
Results
Acute liver injury and repair
following 50% PH
A total of 50 mice were used in the present study.
The 50% PH surgery was performed in 20 mice and the sham operation
in 20 mice, while the 10 remaining mice were used as NC. In each
group, 10 mice were sacrificed 3 days post-operatively and the
other 10 were sacrificed at 14 days post-operatively. Animals from
the NC group were sacrificed at the same time as those of Sham3
group. None of the mice was accidentally dead after the operation.
Serum ALT and AST were tested for each mouse (Fig. 1). The mice in the group sacrificed 3
days post-50% PH (LR3 group) exhibited acute liver injury compared
with those in the group sacrificed 3 days post-sham operation
(Sham3 group) and the NC group, while the liver function of the
mice sacrificed 14 days post-50% PH (LR14 group) had partially
recovered (Fig. 1A). Liver
histology slides of group LR3 exhibited swelling of certain
hepatocytes with acidophilic necrosis and the majority of
hepatocytes had single enlarged, double or multiple nuclei,
prominent nucleoli caryocinesia and vesicular bodies. In the LR14
group, single enlarged or double nuclei were still present but with
less prominent acidophilic bodies in most hepatocytes (Fig. 2).
 | Figure 1Changes in liver function and gut
microbiota following 50% partial hepatectomy. (A) Liver ALT and AST
levels. Values are expressed as the median (interquartile range)
(n=10 per group). *P<0.05, **P<0.01
according to ANOVA with LSD post-hoc test. (B) PCA of the bacterial
community of the small intestinal contents of the mice. (C)
Three-part Venn diagrams display the OTUs shared among the groups.
The numbers in the diagrams represent the numbers of unique OTUs in
each group or shared between groups as their areas intersect. (D)
Unweighted UniFrac distance-based PCoA plots. (E) Rarefaction
curves. (F) Unweighted UniFrac distance-based NMDS. Groups: NC,
normal control; LR3, 3 days post-liver resection; LR14, 14 days
post-liver resection; Sham3, 3 days post-sham operation; Sham14, 14
days post-sham operation; OTU, operational taxonomic unit; PCA,
principal component analysis; PCoA, principal coordinate analysis;
NMDS, non-metric multidimensional scaling; ALT, alanine
aminotransferase; AST, aspartate aminotransferase. |
Overall structural changes of
intestinal microbiota following 50% PH
The V3-V4 region of the 16S rRNA was sequenced on
the Illumina MiSeq platform and a total of 4,050,657 raw reads were
obtained. After multiple paired-ends joining, quality trimming and
filtering steps of the raw sequencing data, a total of 3,738,823
high-quality sequences were acquired, with an average of 74,776
(range, 54,900-93,663) sequences per sample. Specifically, 717,489,
775,830, 711,428, 777,509 and 756,567 sequences were acquired from
the NC, Sham3, Sham14, LR3 and LR14 group, respectively. All of the
sequences were clustered into 5,799 OTUs and aggregated into 42
phyla and 815 genera. There were 1,904 species-level OTUs in the NC
group and 2,575, 1,942, 1,992 and 1,794 OTUs in the Sham3, Sham14,
LR3 and LR14 group, respectively (Table
I). Three-part Venn diagrams were generated to display the
overlaps among groups, demonstrating that 684 of the total 4,387
OTUs were shared among the NC, Sham3 and LR3 groups, 572 out of
3,630 OTUs were shared among the NC, LR3 and LR14 group, and 574
out of 3,899 OTUs were shared among the NC, Sham14 and LR14 groups
(Fig. 1C). Of note, the LR groups
had the lowest number of unique OTUs in their comparisons.
 | Table Iα-Diversity estimation for each group
from the pyrosequencing analysis. |
Table I
α-Diversity estimation for each group
from the pyrosequencing analysis.
| Groups | OTUs (n) | Good coverage
(%) | Chao1 | 95% CI | Observed OTUs | 95% CI | Shannon | Simpson |
|---|
| NC | 1904 | 99.94 | 384 | 298.6-469.0 | 376 | 299-452 | 4.0611 | 0.8071 |
| Sham3 | 2575 | 99.92 | 497 | 409.1-588.2 | 486 | 401-574 | 4.7900 | 0.8681 |
| LR3 | 1992 | 99.95 | 391 | 329.2-466.0 | 384 | 316-456 | 4.2855 | 0.8334 |
| Sham14 | 1942 | 99.96 | 403 | 287.4-516.2 | 397 | 284-518 | 4.5358 | 0.8404 |
| LR14 | 1794 | 99.95 | 362 | 287.6-447.3 | 355 | 281-439 | 4.3405 | 0.8393 |
Microbiota community richness and
diversity changes following 50% PH
Analysis of the α-diversity indicated that the
observed OTUs and species richness (Chao1) were lower in the LR
groups than those in the Sham group but without any significant
difference (Table I). The species
diversity (Shannon and Simpson indices) was also lower in the LR
groups, while that in the LR14 group was slightly higher than that
in the LR3 group (Shannon: 4.3405 vs. 4.2855, P=0.22; Simpson:
0.8393 vs. 0.8334, P=0.48; Table
I). Good coverage of all samples was >99.8% and rarefaction
curves of all the samples approached a plateau (Fig. 1E), indicating sufficient sequencing
for the coverage of all OTUs. The results indicated that after LR,
the species richness was steadily decreased (Chao1 and observed
OTUs) over time. Of note, the trend of the changes in the Sham
group was more obvious than that in the LR group. Furthermore, LR
decreased the species diversity, and conversely, the sham operation
increased it (Table I). We
hypothesize that anesthesia, surgical trauma and stress of the sham
operation could increase the species diversity, while acute liver
injury of the LR could decrease the species diversity.
Furthermore, β-diversity was assessed by PCA, PCoA
and NMDS based on the unweighted UniFrac distances between the
samples (23,24). PCA revealed that the LR groups were
distinguished from the NC group (Fig.
1B). PCoA indicated that most of the samples from the NC, Sham3
and LR3 groups were clustered together, while they were separated
from the Sham14 and LR14 groups (Fig.
1D). NMDS analysis provided a similar result (Fig. 1F).
Alterations of taxonomy community at
the phylum level following 50% PH
The 16S rRNA amplicon libraries from the small
intestinal contents were sequenced and established to characterize
the alterations in gut microbiota associated with 50% PH-induced
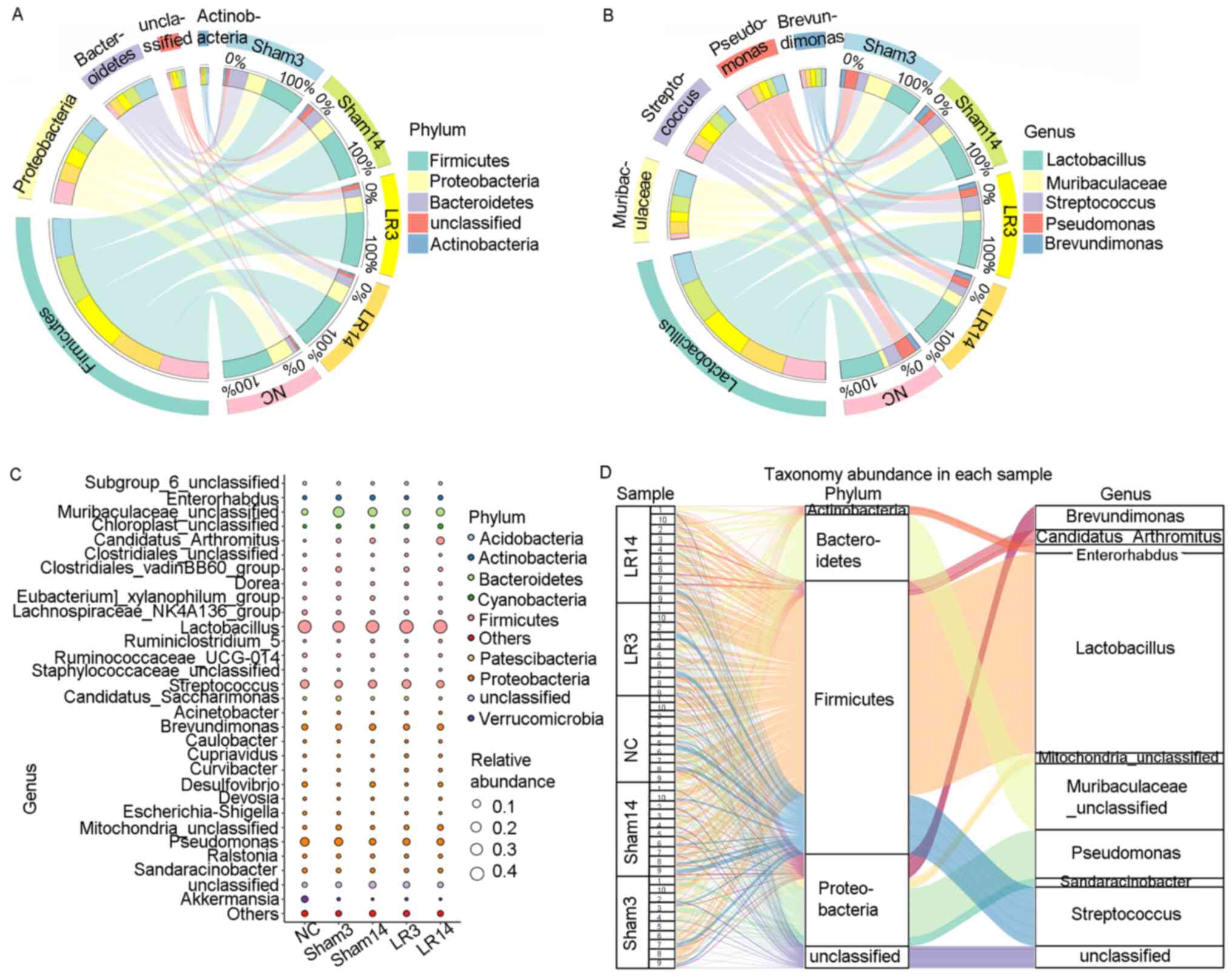
acute liver injury and repair. At the phylum level, the 5 dominant
bacterial phyla were Firmicutes, Proteobacteria, Bacteroidetes,
Actinobacteria and Cyanobacteria, accounting for >90% of all
sequences in all groups (Figs. 3
and 4). Relative abundances of the
intestinal microbiota at the bacterial phylum level among different
groups were presented in a hierarchical clustering heatmap
(Fig. 3A). LR led to an increase in
Firmicute abundance but decreases in Bacteroidetes and
Actinobacteria, compared with the Sham group, but without any
statistically significant difference (Figs. 3A and B and 4A
and C). In group LR3, significant
Cyanobacteria enrichment and Fusobacteria depletion in comparison
with the NC and Sham groups was observed (P=0.03 and 0.02,
respectively; Fig. 5A). Then in the
liver repair phase (LR14), Cyanobacteria were enriched and
Verrucomicrobia were depleted compared with the NC and Sham groups
(P=0.04 and <0.001, respectively; Fig. 5A). Furthermore, compared with those
in the LR3 group, mice in the LR14 group exhibited a decrease in
Firmicute abundance and increases in Proteobacteria, Bacteroidetes,
Actinobacteria and Cyanobacteria, but without any statistical
significance (Figs. 3A and B and 4A
and C). In the LR14 group,
significantly depleted Verrucomicrobia and Chloroflexi abundance
was observed in comparison to the LR3 group (P=0.01 and 0.03,
respectively; Fig. 5A).
 | Figure 5Box plots displaying differences in
the gut microbiota following 50% partial hepatectomy at the
bacterial (A) phylum level and (B) genus level. Values are
expressed as the median (interquartile range) (n=10 per group).
#P<0.05, ##P<0.01, nonparametric
Mann-Whitney U-test; *P<0.05, **P<0.01,
nonparametric Kruskal-Wallis H-test, the statistical significance
is indicated among all three groups compared. Groups: NC, normal
control; LR3, 3 days post-liver resection; LR14, 14 days post-liver
resection; Sham3, 3 days post-sham operation; Sham14, 14 days
post-sham operation. |
Alterations of taxonomy community at
the genus level following 50% PH
At the genus level, the 5 most abundant bacterial
genera were distributed in phyla as follows: Two Firmicutes
(Lactobacillus and Streptococcus), one Bacteroidete
(Muribaculaceae) and two Proteobacteria (Pseudomonas
and Brevundimonas) (Fig.
4B-D). The remaining 4 of the 9 most abundant bacterial genera
were Candidatus arthromitus, Mitochondria,
Sandaracinobacter and Enterorhabdus, which belong to
the phyla Firmicutes, Proteobacteria, Proteobacteria and
Actinobacteria, respectively (Figs.
3C and 4C and D). In the LR3 group, significant increases
in Chloroplast, Curvibacter, Pelomonas,
Ruminococcaceae UCG-005 and Blautia (belonging to the
phyla Cyanobacteria, Proteobacteria, Proteobacteria, Firmicutes and
Firmicutes, respectively) but a sharp decrease in
Akkermansia and Eubacterium coprostanoligenes
(belonging to the phyla Verrucomicrobia and Firmicutes,
respectively) were observed compared with the Sham and NC groups
(P<0.05; Fig. 5B). In the liver
repair phase (LR14 group), Candidatus arthromitus,
Chloroplast and Kineococcus (belonging to the phyla
Firmicutes, Cyanobacteria and Actinobacteria, respectively) were
enriched, while Escherichia-Shigella and
Faecalibacterium (belonging to the phyla Proteobacteria and
Firmicutes, respectively) were depleted compared with the Sham and
NC groups (P<0.05; Fig. 5B).
Compared with those in group LR3, mice in group LR14 exhibited
significant increases in Candidatus arthromitus and
Mucispirillum (belonging to the phyla Firmicutes and
Deferribacteres, respectively) but significant decreases in
Ruminococcus, Subdoligranulum, Romboutsia,
Clostridium sensu stricto 1 and Paenibacillus
(phylum, Firmicutes), Bifidobacterium,
Corynebacterium and Streptomyces (phylum,
Actinobacteria), Nevskia and Escherichia-Shigella
(phylum, Proteobacteria), Chryseobacterium and
Parabacteroides (phylum, Bacteroidetes) and
Akkermansia (phylum, Verrucomicrobia) were obtained
(P<0.05; Fig. 5B).
Specific bacterial taxa with
significant changes in relative abundance following 50% PH
LEfSe analysis was used to distinguish more specific
bacterial taxa whose relative abundance was significantly different
among the NC, Sham and LR groups (25), revealing that in the LR3 group, the
microbiota had an increased abundance in Oxyphotobacteria at the
class level, Chloroplast at the order level, Chloroplast at the
family level, Chloroplast and Curvibacter at the
genus level and Chloroplast sp. and Curvibacter sp.
at the species level (distinct from NC and Sham, LDA scores >3;
Fig. 6A). This analysis also
revealed that in the LR14 group, the microbiota was enriched in
Cyanobacteria at the phylum level, Oxyphotobacteria at the class
level, Chloroplast at the order level, Clostridiaceae 1,
Chloroplast and Rhodanobacteraceae at the family level,
Candidatus arthromitus, Chloroplast and
Tahibacter at the genus level and Candidatus
arthromitus sp., Chloroplast sp. and Tahibacter
sp. at the species level (distinct from NC and Sham, LDA scores
>3; Fig. 6B). Furthermore,
Fig. 6C provides the differential
taxa between the LR3 and LR14 groups determined using the LEfSe
analysis method, revealing that in the LR14 group, the microbiota
exhibited an increased abundance in Clostridiaceae 1 at the family
level, Candidatus arthromitus, Eubacterium nodatum and
Ruminococcaceae NK4A214 at the genus level and Candidatus
arthromitus sp., Eubacterium nodatum sp.,
Papillibacter sp. and Ruminococcaceae NK4A214 sp. at
the species level (distinct from LR3, LDA scores >3; Fig. 6C). Collectively, all of these
results demonstrated that the gut microbiota may have an important
role in the acute liver injury and repair following 50% PH.
Potential functional changes of the
gut microbiome following 50% PH
The metagenome Kyoto Encyclopedia of Genes and
Genomes (KEGG) orthologies of the 50 samples were predicted with
PICRUSt software based on the Greengenes database (26). Then KEGG orthologies were collapsed
to the pathway level (KEGG level 3) by PICRUSt. The differences in
KEGG pathways between groups were determined by STAMP (27). Fig.
7 provides the statistically significant KEGG pathways compared
between pairs of groups (LR3 vs. Sham3, LR14 vs. Sham14 and LR3 vs.
LR14). In group LR3, a significant enhancement of the ‘myo-, chiro-
and scillo-inositol degradation’ and ‘isoprene biosynthesis II
(engineered)’ pathways, but a significant decline of the
‘incomplete reductive TCA cycle’ pathway, compared to the Sham3
group was obtained (Fig. 7A).
However, in group LR14, enhancement of ‘anhydromuropeptides
recycling’, ‘GDP-mannose biosynthesis’ and ‘urea cycle’ pathways,
but a decline of the ‘superpathway of L-isoleucine biosynthesis I’
pathway in comparison with the Sham14 group was present (Fig. 7B). Compared to the LR3 group, the
LR14 group exhibited significant enhancements in the ‘GDP-mannose
biosynthesis’ and ‘urea cycle’ pathways but decreases in numerous
other pathways, including ‘cob(II)yrinate a,c-diamide biosynthesis
I (early cobalt insertion)’, ‘superpathway of (Kdo)2-lipid A
biosynthesis’, ‘superpathway of L-tryptophan biosynthesis’,
‘enterobactin biosynthesis’ and ‘superpathway of chorismate
metabolism’ (Fig. 7C).
Discussion
The intestinal microecology has critical roles in
regulating metabolic processes (7,8),
modulating immunity (9,10) and protecting the host against
pathogenic microbes (11) and is
considered an ignored endocrine organ (1,7,8).
Signals from the intestinal microbiota have been related to the
maintenance of healthy host function and prevention of various
diseases, including obesity, diabetes, inflammatory bowel disease,
liver disease, allergy and cancer (1-14).
Studies have revealed that changes in the intestinal
microbiota have an important role in the development of liver
diseases (1-6,13).
The diversity, richness and function of the intestinal microbiota
have been associated with hepatitis B, liver cirrhosis and acute
liver injury (1-6).
The intestinal microbiota was determined to influence the host
immune response to hepatitis B virus and may determine whether
acute or chronic hepatitis B occurs (2). Patients with chronic hepatitis B and
cirrhosis exhibited significantly increased Enterococcus and
Enterobacteriaceae levels, but markedly decreased
Bifidobacteria and Lactobacillus levels compared with
healthy subjects (2,3). To a certain extent, liver cirrhosis
may be caused by bacterial products from the intestine (3,4).
Overgrowth of harmful bacteria in the intestine leads to increased
mucosal permeability, which causes bacterial or endotoxin
translocation and thus activate the liver's innate immune system
(1,2,13). The
lipopolysaccharide-toll-like receptor (TLR) 4 pathway and the
unmethylated CpG DNA-TLR9 pathway may be 2 important related immune
mechanisms (2). Serum lipids
(phospholipids, free fatty acids, eicosapentaenoic acid,
arachidonic acid and docosahexaenoic acid) were indicated to have
significant correlations with Enterococcus,
Enterobacteriaceae, Bifidobacterium,
Bacteroides, Lactobacillus and Candida
(5). These correlations differed
among normal liver, liver fibrosis and liver cirrhosis groups,
suggesting that chronic liver disease itself alters the intestinal
flora-associated fatty acid metabolism (5). The intestinal microbiota produces a
variety of compounds that have critical roles in regulating the
activity of the liver (1,2). Studies on CCl4-induced
acute liver injury revealed that Firmicute abundance increased and
Bacteroidetes decreased over time, and initial alterations in the
Firmicute/Bacteroidete ratio may be beneficial to liver repair
(6). However, the changes of the
intestinal microbiota during PH-induced acute liver injury had
remained elusive. In order to simulate the clinical situation as
much as possible, a 50% PH model was selected for the present
study. Regardless of the type of liver damage, the general
association between liver damage and changes in the microbiota was
demonstrated in the present study.
In the present study, changes in the intestinal
microbiota following 50% PH-induced acute liver injury and repair
were assessed. α-Diversity analysis suggested that LR induced a
reduction of observed OTUs, species richness (Chao1) and species
diversity (Shannon and Simpson indices), although without any
significant differences. An increase in Firmicute abundance and
decreases in Bacteroidetes and Actinobacteria were detected in
association with LR and alterations of the Firmicute/Bacteroidete
ratio were indicative of diverse disease processes (28). LR significantly induced
Cyanobacteria enrichment over time. In contrast to acute liver
injury, liver repair led to a significant depletion of
Verrucomicrobia, Chloroflexi and Deferribacteres, suggesting that
these bacterial phyla may also have indispensable roles in 50% PH.
In addition, at the genus level, LR3 led to significant
upregulation of Chloroplast, Curvibacter,
Pelomonas, Ruminococcaceae UCG-005 and Blautia
but downregulation of Akkermansia and Eubacterium
coprostanoligenes compared with the Sham and NC groups.
Obviously, acute liver injury caused overgrowth of intestinal
pathogenic microorganisms and the fading of intestinal beneficial
bacteria. The differences in bacterial genera between the LR14 and
LR3 groups suggested that in the liver repair phase, the amount of
pathogenic microorganisms decreased, while that of beneficial
bacteria increased, resulting in the restoration of the intestinal
microecological balance.
Among the significantly different bacterial taxa,
previous studies also revealed that Ruminococcaceae exhibited a
higher transcriptional activity than abundance in ulcerative
colitis patients (14) and also
require anaerobic conditions and carbohydrate energy sources from
dietary fibre (29). Cyanobacteria
and chloroplasts were responsible for pathways related to
photosynthesis (29).
Clostridium, known as pathogenic bacteria (30), have an important role in mice with
obesity (31,32). Akkermansia, regarded as
mucus-degrading bacteria, which were able to adhere to the gut
epithelium and reinforce enterocyte layer integrity in vitro
(33) and initiate mucus
degradation to generate oligosaccharides and acetate, evoking
colonized bacterial growth and resistance to pathogenic
microorganisms in vivo, were beneficial for the composition
of the mucus-related microbiota (34). An increased Akkermansia
population in the gut microbiota was associated with the
improvement of metabolic syndrome and prevention of diet-induced
obesity, insulin resistance and intestinal inflammation in high-fat
diet-fed mice (35).
Functional KEGG pathway analysis provided the
specific and distinct pathways of the gut microbiome associated
with acute liver injury and repair following 50% PH, revealing that
the potential functions of the gut microbiota differed
significantly during the liver injury and repair phase. Acute liver
injury led to the upregulation of biosynthesis of isoprene, heme,
geranylgeranyldiphosphate and factor 420, and enhancement of
degradation of N-acetylneuraminate, myo-inositol and myo-, chiro-
and scillo-inositol, associated with increased Chloroplast,
Curvibacter, Pelomonas, Ruminococcaceae
UCG-005 and Blautia abundance as compared to the Sham3
group. Furthermore, acute liver injury induced downregulation of
glycolysis V, formaldehyde assimilation II, biosynthesis of
thiazolet, TCA cycle V and incomplete reductive TCA cycle,
associated with decreased Akkermansia and Eubacterium
coprostanoligenes abundance as compared to the Sham3 group.
Compared with the LR3 group, mice in the liver repair phase (LR14
group) exhibited upregulation of GDP-mannose biosynthesis and the
urea cycle, associated with increased Candidatus arthromitus
and Mucispirillum abundance, as well as downregulation of
the pathway of chorismate metabolism and biosynthesis of
cob(II)yrinate a,c-diamide, (Kdo)2-lipid A, L-tryptophan and
enterobactin. The present study thus revealed the alterations of
signal transduction, transcription, cell motility, as well as
metabolism of amino acids, lipids, glucose, cofactors and
terpenoids, and xenobiotics pathways associated with 50% PH-induced
acute liver injury and repair.
These results suggested that acute liver injury may
cause microecological dysbiosis, affecting the potential function
of the intestinal microbiota (6).
At the same time, liver injury induced a compensatory mechanism in
the host and the gut microbiota may react to the liver and
influence liver repair through changes in microecological function.
However, it remains elusive how the intestinal microbiota may be
modified to promote the recovery of liver function following
hepatectomy. This is a major limitation of the present study and
provides a direction for further research.
In conclusion, in the present study, the structural
and functional changes of the gut microbiota following 50% PH were
investigated. It was indicated that 50% PH reduced the species
richness and diversity, caused dysbiosis of the microbiota and
altered signal transduction, transcription, cell motility and
metabolism of amino acids, lipids and glucose pathways of the
intestinal microbiota. A further understanding of the
gut-microbiota-liver metabolic network may provide novel strategies
for the recovery of patients undergoing liver surgery.
Acknowledgements
The authors would like to thank Mrs. Lei Chen
(Medical Information Department of The First Affiliated Hospital,
School of Medicine, Zhejiang University), Miss Li Liu (Library of
The First Affiliated Hospital, School of Medicine, Zhejiang
University) and Mr. Zhuo Yang (LC-Bio Technology Co., Ltd.,
Hangzhou, China) for their help with data processing, and Dr Han
Zhang (Department of Pathology, The First Affiliated Hospital,
School of Medicine, Zhejiang University) for his help with the
liver histology.
Funding
Funding: The present study was supported by the Foundation for
Innovative Research Groups of the National Natural Science
Foundation of China (grant no. 81721091), the National Natural
Science Foundation of China (grant no. 81770645) and the Basic
Public Welfare Research Program of Zhejiang Province (grant no.
LGF18H030006).
Availability of data and materials
The raw sequencing data and sample information of
this study have been submitted to the NCBI Sequence Read Archive
under the BioProject accession no. PRJNA678005 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA678005).
Authors' contributions
YS, ZHH and SSZ conceived and designed the study.
YS, YCJ and HL performed the animal experiments. YCJ and HL
performed the biochemical analysis and liver histology. FZ, YCJ and
HL performed the microecology analysis. YS and YCJ did the data
analysis. YS drafted the manuscript. YS and HL confirmed the
authenticity of the raw data. SSZ and ZHH supervised the study. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
The Animal Experimental Ethics Committee of the
First Affiliated Hospital, School of Medicine, Zhejiang University
(Hangzhou, China) approved this study (approval no. 2019-1086).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Usami M, Miyoshi M and Yamashita H: Gut
microbiota and host metabolism in liver cirrhosis. World J
Gastroenterol. 21:11597–11608. 2015.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Yang R, Xu Y, Dai Z, Lin X and Wang H: The
immunologic role of gut microbiota in patients with chronic HBV
infection. J Immunol Res. 2018(2361963)2018.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Lu H, Wu Z, Xu W, Yang J, Chen Y and Li L:
Intestinal microbiota was assessed in cirrhotic patients with
hepatitis B virus infection. Intestinal microbiota of HBV cirrhotic
patients. Microb Ecol. 61:693–703. 2011.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Jun DW, Kim KT, Lee OY, Chae JD, Son BK,
Kim SH, Jo YJ and Park YS: Association between small intestinal
bacterial overgrowth and peripheral bacterial DNA in cirrhotic
patients. Dig Dis Sci. 55:1465–1471. 2010.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Usami M, Miyoshi M, Kanbara Y, Aoyama M,
Sakaki H, Shuno K, Hirata K, Takahashi M, Ueno K, Hamada Y, et al:
Analysis of fecal microbiota, organic acids and plasma lipids in
hepatic cancer patients with or without liver cirrhosis. Clin Nutr.
32:444–451. 2013.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Dong X, Feng X, Liu J, Xu Y, Pan Q, Ling
Z, Yu J, Yang J, Li L and Cao H: Characteristics of intestinal
microecology during mesenchymal stem cell-based therapy for mouse
acute liver injury. Stem Cells Int. 2019(2403793)2019.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Tremaroli V and Bäckhed F: Functional
interactions between the gut microbiota and host metabolism.
Nature. 489:242–249. 2012.
|
|
8
|
Cani PD: Metabolism in 2013: The gut
microbiota manages host metabolism. Nat Rev Endocrinol. 10:74–76.
2014.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Maynard CL, Elson CO, Hatton RD and Weaver
CT: Reciprocal interactions of the intestinal microbiota and immune
system. Nature. 489:231–241. 2012.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Viaud S, Saccheri F, Mignot G, Yamazaki T,
Daillère R, Hannani D, Enot DP, Pfirschke C, Engblom C, Pittet MJ,
et al: The intestinal microbiota modulates the anticancer immune
effects of cyclophosphamide. Science. 342:971–976. 2013.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Fukuda S, Toh H, Hase K, Oshima K,
Nakanishi Y, Yoshimura K, Tobe T, Clarke JM, Topping DL, Suzuki T,
et al: Bifidobacteria can protect from enteropathogenic infection
through production of acetate. Nature. 469:543–547. 2011.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Schroeder BO and Bäckhed F: Signals from
the gut microbiota to distant organs in physiology and disease. Nat
Med. 22:1079–1089. 2016.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Lin R, Zhou L, Zhang J and Wang B:
Abnormal intestinal permeability and microbiota in patients with
autoimmune hepatitis. Int J Clin Exp Pathol. 8:5153–5160.
2015.PubMed/NCBI
|
|
14
|
Moen AEF, Lindstrøm JC, Tannæs TM, Vatn S,
Ricanek P, Vatn MH and Jahnsen J: IBD-Character Consortium. The
prevalence and transcriptional activity of the mucosal microbiota
of ulcerative colitis patients. Sci Rep. 8(17278)2018.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Michalopoulos GK: Liver regeneration. J
Cell Physiol. 213:286–300. 2007.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Michalopoulos GK: Hepatostat: Liver
regeneration and normal liver tissue maintenance. Hepatology.
65:1384–1392. 2017.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Flye MW and Yu S: Augmentation of
cell-mediated cytotoxicity following 50% partial hepatectomy.
Transplantation. 49:581–587. 1990.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Cao H, Yu J, Xu W, Jia X, Yang J, Pan Q,
Zhang Q, Sheng G, Li J, Pan X, et al: Proteomic analysis of
regenerating mouse liver following 50% partial hepatectomy.
Proteome Sci. 7(48)2009.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Brandt HH, Nißler V and Croner RS: The
influence of liver resection on intrahepatic tumor growth. J Vis
Exp. 110(e53946)2016.PubMed/NCBI View
Article : Google Scholar
|
|
20
|
Fadrosh DW, Ma B, Gajer P, Sengamalay N,
Ott S, Brotman RM and Ravel J: An improved dual-indexing approach
for multiplexed 16S rRNA gene sequencing on the Illumina MiSeq
platform. Microbiome. 2(6)2014.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Callahan BJ, McMurdie PJ, Rosen MJ, Han
AW, Johnson AJ and Holmes SP: DADA2: High-resolution sample
inference from Illumina amplicon data. Nat Methods. 13:581–583.
2016.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Blaxter M, Mann J, Chapman T, Thomas F,
Whitton C, Floyd R and Abebe E: Defining operational taxonomic
units using DNA barcode data. Philos Trans R Soc Lond B Biol Sci.
360:1935–1943. 2005.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Bolyen E, Rideout JR, Dillon MR, Bokulich
NA, Abnet CC, Al-Ghalith GA, Alexander H, Alm EJ, Arumugam M,
Asnicar F, et al: Author correction: Reproducible, interactive,
scalable and extensible microbiome data science using QIIME 2. Nat
Biotechnol. 37(1091)2019.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Caporaso JG, Kuczynski J, Stombaugh J,
Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich
JK, Gordon JI, et al: QIIME allows analysis of high-throughput
community sequencing data. Nat Methods. 7:335–336. 2010.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Segata N, Izard J, Waldron L, Gevers D,
Miropolsky L, Garrett WS and Huttenhower C: Metagenomic biomarker
discovery and explanation. Genome Biol. 12(R60)2011.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Langille MG, Zaneveld J, Caporaso JG,
McDonald D, Knights D, Reyes JA, Clemente JC, Burkepile DE, Vega
Thurber RL, Knight R, et al: Predictive functional profiling of
microbial communities using 16S rRNA marker gene sequences. Nat
Biotechnol. 31:814–821. 2013.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Parks DH, Tyson GW, Hugenholtz P and Beiko
RG: STAMP: Statistical analysis of taxonomic and functional
profiles. Bioinformatics. 30:3123–3124. 2014.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Liu HX, Rocha CS, Dandekar S and Wan YJ:
Functional analysis of the relationship between intestinal
microbiota and the expression of hepatic genes and pathways during
the course of liver regeneration. J Hepatol. 64:641–650.
2016.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Crespo-Piazuelo D, Estellé J, Revilla M,
Criado-Mesas L, Ramayo-Caldas Y, Óvilo C, Fernández AI, Ballester M
and Folch JM: Characterization of bacterial microbiota compositions
along the intestinal tract in pigs and their interactions and
functions. Sci Rep. 8(12727)2018.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Samul D, Worsztynowicz P, Leja K and
Grajek W: Beneficial and harmful roles of bacteria from the
Clostridium genus. Acta Biochim Pol. 60:515–521. 2013.PubMed/NCBI
|
|
31
|
Masumoto S, Terao A, Yamamoto Y, Mukai T,
Miura T and Shoji T: Non-absorbable apple procyanidins prevent
obesity associated with gut microbial and metabolomic changes. Sci
Rep. 6(31208)2016.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Jiao X, Wang Y, Lin Y, Lang Y, Li E, Zhang
X, Zhang Q, Feng Y, Meng X and Li B: Blueberry polyphenols extract
as a potential prebiotic with anti-obesity effects on C57BL/6 J
mice by modulating the gut microbiota. J Nutr Biochem. 64:88–100.
2019.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Reunanen J, Kainulainen V, Huuskonen L,
Ottman N, Belzer C, Huhtinen H, de Vos WM and Satokari R:
Akkermansia muciniphila adheres to enterocytes and strengthens the
integrity of the epithelial cell layer. Appl Environ Microbiol.
81:3655–3662. 2015.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Belzer C and de Vos WM: Microbes
inside-from diversity to function: The case of Akkermansia. ISME J.
6:1449–1458. 2012.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Anhê FF, Roy D, Pilon G, Dudonné S,
Matamoros S, Varin TV, Garofalo C, Moine Q, Desjardins Y, Levy E
and Marette A: A polyphenol-rich cranberry extract protects from
diet-induced obesity, insulin resistance and intestinal
inflammation in association with increased Akkermansia spp.
population in the gut microbiota of mice. Gut. 64:872–883.
2015.PubMed/NCBI View Article : Google Scholar
|