Introduction
The liver performs and regulates numerous
physiological functions, and has the ability to regenerate
(1,2). It is an important line of defense
against microorganisms and is also a target of the dysregulated
inflammation induced by infectious diseases of the liver such as
steatohepatitis (3). Based on the
International Classification of Diseases, Ninth Revision, Clinical
Modification, severe sepsis and septic shock are main causes for
hospital patients to be admitted to intensive care unit (4). Sepsis is defined as a
life-threatening organ dysfunction caused by a dysregulated host
response to infection (5). Among
the organ failures involved in sepsis, hepatic dysfunction has a
high level of prognostic relevance for the course of sepsis and is
a powerful independent predictor of mortality (6).
The Periplaneta americana (PA) extract is
found in the American cockroach and comprises polyols, peptides,
epidermal growth factors, sticky sugar acid, amino acids and
further active components (7). The
PA extract notably alleviates swelling of liver tissue and stomach
pain in gastric ulcer (8,9). Furthermore, it is widely used in
China for its beneficial effects on blood circulation, hematocele
clearance and digestion (10).
Xinmailong (XML), which is typically made from PA, reverses the
cardiomyocyte damage caused by lipopolysaccharide (LPS) by
mediating mitophagy and downregulating the expression of
inflammatory factors (11). PA
extract protected the intestinal mucosal barrier in patients with
sepsis, thereby improving the physical condition of the patients
and ameliorating prognosis (7). PA
extract reduced the level of endotoxin, attenuated gastrointestinal
complications, and improved immune function and nutritional status
in patients with systemic inflammatory response syndrome (7,12).
The results of a previous study demonstrated that XML alleviated
intestinal inflammation and improved the intestinal mucosal barrier
function (10). By decreasing the
expression levels of fibrosis-related genes, the liver function of
mice was enhanced, and the pathological fibrosis associated with
liver were mitigated or even reversed, resulting in inhibition of
liver fibrosis progression (13).
Additionally, PA extract accelerated liver regeneration via a
complex network of pathways, including AMP-activated protein kinase
(AMPK) signaling (14). The
activation of AMPK maintained mitochondrial function and regulated
autophagy, thus preventing sepsis-induced cell and organ injury
(15). A previous study has
proposed that inhibition of the AMPK/proliferator-activated
receptor γ coactivator-1α (PGC-1α) signaling pathway in rat liver
induced diseases associated with mitochondrial dynamics (16).
The present study aimed to investigate the role of
XML in LPS-induced liver injury and determined whether the
potential underlying mechanism is associated with regulation of the
AMPK/PGC-1α signaling pathway.
Materials and methods
Cell culture and treatments
The AML12 cell line (cat. no. 3101MOUSCSP550) was
purchased from The Cell Bank of Type Culture Collection of the
Chinese Academy of Sciences and maintained in DMEM (Invitrogen;
Thermo Fisher Scientific, Inc.) supplemented with 10% FBS
(Sigma-Aldrich; Merck KGaA), 100 U/ml penicillin and 100 µg/ml
streptomycin (Beyotime Institute of Biotechnology). Once cells
reached 80% confluence, they were treated with 100 ng/ml LPS for 12
h at 37˚C. Certain cells were also preincubated with 0.25, 0.5, 1,
2 and 4 mg/ml of the PA extract, XML (Yunnan Tengyao
Pharmaceutical, Co., Ltd.) for 48 h. Cells were treated with 2
mg/ml AMPK inhibitor, compound C (Sigma-Aldrich; Merck KGaA) for 1
h at 37˚C before model establishment.
Detection of cytochrome c
Cytochrome c content in the mitochondria and
cytoplasm was detected using a Quantikine Mouse Cytochrome c
Immunoassay kit obtained from R&D Systems, Inc. (cat. no.
MCTC0), according to the manufacturer's protocol. The AML12 cells
were lysed with lysis buffer (Beyotime Institute of Biotechnology)
containing protease inhibitors (Sigma-Aldrich; Merck KGaA). The
lysate was centrifuged for 10 min at 750 x g, and the supernatants
were collected and subsequently centrifuged at 15,000 x g for 15
min at 4˚C. The supernatants were used as the cytosolic fractions.
The pellet containing the mitochondria was combined with PBS plus
0.5% Triton X-100 (Sigma-Aldrich; Merck KGaA) for 10 min at 4˚C.
The pellet was centrifuged at 15,000 x g, 4˚C for 10 min, and the
supernatant was used as the mitochondrial fraction.
Luciferin-luciferase reaction
Mitochondria were collected from cells by
centrifugation at 4˚C, 12,000 x g for 8 min. Then, 100 µl sample
was added in a 96-well plate before 100 µl ATP reagent was added
for 2 min. ATP concentration was measured using an enhanced ATP
assay kit (cat. no. S0027, Beyotime Institute of Biotechnology),
according to the manufacturer's protocol.
MTT assay
Cell viability was assessed using an MTT assay.
Cells were seeded into 96-well plates at a density of
2x104 cells/well following culture with or without XML
at concentrations of 0.25, 0.5, 1, 2 and 4 mg/ml respectively for
48 h at 37˚C. The DMEM containing serum was replaced with MTT
(Sigma-Aldrich; Merck KGaA) following the corresponding treatment.
Subsequently, 0.5 mg/ml MTT reagent was added to each well and
incubated for 4 h at 37˚C. The cell supernatant was discarded and
replaced with DMSO (150 µl/well). The absorbance was measured at a
wavelength of 570 nm using a microplate reader (Bio-Rad
Laboratories, Inc.).
ELISA
AML12 cells were seeded into 96-well plates at a
density of 1x105 cells/well and co-treated with 100
ng/ml LPS, 4 mg/ml XML and 5 mM compound C for 12 h. The
supernatants of the cultured cells were collected in a centrifuge
at 1,000 x g, 4˚C for 15 min to measure the secretion levels of
IL-1β (cat. no. F10770), IL-6 (cat. no. F10830) and TNF-α (cat. no.
F11630) using commercial ELISA kits, according to the
manufacturer's protocol (Shanghai Xitang Biological Technology Co.,
Ltd.).
Measurement of oxidative
stress-related markers
The cell malondialdehyde (MDA; cat. no. A003-4-1)
assay kit and total superoxide dismutase (SOD; cat. no. A001-1-2)
assay kit were purchased from Nanjing Jiancheng Bioengineering
Institute. The measurement of MDA and SOD was performed according
to the manufacturer's protocol.
TUNEL assay
Apoptosis of AML12 cells was detected using a TUNEL
assay kit (Beyotime Institute of Biotechnology), according to the
manufacturer's protocol. Briefly, 2x105 cells were
washed twice with buffer in 24-well plates and then fixed in 0.5 ml
4% paraformaldehyde for 30 min at 37˚C. Subsequently, cells were
incubated with 50 µl TUNEL reaction buffer for 1 h at 37˚C. The
nuclei were counterstained with 1 µg/ml DAPI in the dark at room
temperature for 15 min. Images were visualized and captured using a
fluorescence microscope (magnification, x200; Olympus Corporation)
and cells were counted in five randomly-selected microscopic
fields.
Reverse transcription-quantitative
(RT-q)PCR
Total RNA was isolated from AML12 cells using
TRIzol® reagent (Thermo Fisher Scientific, Inc.). Total
RNA was reverse transcribed into cDNA using a
SuperScript™ IV First-Strand Synthesis system
(Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturers' protocol. qPCR was subsequently performed using the
SYBR Green PCR system and SYBR Green reagents (Bio-Rad
Laboratories, Inc.) in an ABI 7500 thermal cycler (Applied
Biosystems; Thermo Fisher Scientific, Inc.). The following
thermocycling conditions were used for qPCR: Initial denaturation
at 95˚C for 3 min, followed by 40 cycles of denaturation at 95˚C
for 10 sec, annealing at 60˚C for 5 sec and extension at 72˚C for
10 sec. The sequences of the primers were as follows: NRF1 forward,
5'-CGCAGCACCTTTGGAGAA-3' and reverse, 5'-CCCGACCTGTGGAATACTTG-3';
TFAM forward, 5'-GGAATGTGGAGCGTGCTAAA-3' and reverse,
5'-TGCTGGAAAAACACTTCGGAATA-3'; TNF-α forward,
5'-CGTGCTCCTCACCCACAC-3' and reverse, 5'-GGGTTCATACCAGGGTTTGA-3';
IL-6 forward, 5'-ACAACCACGGCCTTCCCTACTT-3' and reverse,
5'-GTGTAATTAAGCCTCCGACT-3'; IL-1β forward,
5'-TCCAGGATGAGGACATGAGCAC-3' and reverse,
5'-GAACGTCACACACCAGCAGGTA-3' and GAPDH forward,
5'-TCACCACCATGGAGAAGGC-3' and reverse, 5'-GCTAAGCAGTTGGTGGTGCA-3'.
GAPDH was used for normalization of mRNA levels. Relative mRNA
expression levels were calculated using the 2-ΔΔCq
method (17).
Western blotting
Following treatment, protein samples were extracted
from cells using RIPA lysis buffer (Beyotime Institute of
Biotechnology) for western blot analysis. A BCA Protein Assay kit
(Beyotime Institute of Biotechnology) was used to determine total
protein concentrations. Proteins (40 µg/lane) were separated via 10
or 12.5% SDS-PAGE, transferred onto PVDF membranes (MilliporeSigma)
and blocked with 5% non-fat milk for 2 h at room temperature. The
membranes were washed three times with 1% TBS-Tween-20 and
incubated with primary antibodies overnight at 4˚C. Following the
primary incubation, membranes were incubated with goat anti-rabbit
horseradish peroxidase (HRP)-conjugated secondary antibody (cat.
no. 7074S, 1:2,000; Cell Signaling Technology, Inc.) or horse
anti-mouse HRP-conjugated secondary antibody (cat. no. 7076S,
1:2,000; Cell Signaling Technology, Inc.) for 1 h at room
temperature. Protein bands were developed using an Enhanced
Chemiluminescence Western Blotting Substrate kit (Thermo Fisher
Scientific, Inc.). The primary antibodies used were as follows:
Anti-phosphorylated (p)-AMPK (cat. no. 2535T, 1:1,000; Cell
Signaling Technology, Inc.); anti-AMPK (cat. no. 5831T, 1:1,000;
Cell Signaling Technology, Inc.); anti-PGC-1α (cat. no. 2178S,
1:1,000; Cell Signaling Technology, Inc.); anti-Bcl-2 (cat. no.
3498T, 1:1,000; Cell Signaling Technology, Inc.); anti-cleaved
caspase-3 (cat. no. 9664T, 1:1,000; Cell Signaling Technology,
Inc.); anti-caspase-3 (cat. no. 14220T, 1:1,000; Cell Signaling
Technology, Inc.); anti-cleaved caspase-9 (cat. no. 20750S,
1:1,000; Cell Signaling Technology, Inc.); anti-caspase-9 (cat. no.
9508T, 1:1,000; Cell Signaling Technology, Inc.); anti-Bax (cat.
no. ab182733, 1:2,000; Abcam) and anti-GAPDH (cat. no. 5174T,
1:1,000; Cell Signaling Technology, Inc.). The grayscale values of
the membranes were semi-quantified using ImageJ software (version
1.52r; National Institutes of Health). Protein levels were
normalized to GAPDH.
Bioinformatics
Kyoto Encyclopedia of Genes and Genomes was used to
analyze the signalling pathways involved in AMPK (genome.ad.jp/kegg/).
Statistical analysis
Statistical analysis was performed using GraphPad
Prism (version 8.0.1; GraphPad Software, Inc.). All experiments
were repeated three times and data are presented as the mean ±
standard deviation. An unpaired Student's t-test was used to
compare differences between two groups, while one-way analysis of
variance (ANOVA) followed by Tukey's post hoc test was used to
compare differences between multiple groups. P<0.05 was
considered to indicate a statistically significant difference.
Results
LPS induces mitochondrial dysfunction
in AML12 cells
To establish the sepsis-induced liver injury model,
LPS was used to induce AML12 cells. As presented in Fig. 1A and B, the level of mitochondrial cytochrome
c significantly decreased compared with the control group,
while that of cytosolic cytochrome c significantly
increased, following LPS induction. Furthermore, ATP synthesis
significantly decreased following LPS stimulation compared with the
untreated group (Fig. 1C). The
relative mRNA expression levels of nuclear respiratory factor 1
(NRF1) and transcription factor A, mitochondrial (TFAM) were
detected via RT-qPCR analysis, and their expression levels were
significantly decreased following treatment of AML12 cells with LPS
compared with the group without LPS (Fig. 1D and E). Oxidative stress and reduction in ATP
production are involved in mitochondrial dysfunction, and along
with apoptosis, are key factors for the occurrence of acute liver
injury caused by sepsis (18).
Collectively, these results suggested that LPS induced
mitochondrial dysfunction in AML12 cells.
XML treatment activates the
AMPK/PGC-1α signaling pathway
Kyoto Encyclopedia of Genes and Genomes analysis
demonstrated that mitochondrial function can be mediated by the
AMPK/PGC-1α signaling pathway; thus, the effect of XML on this
pathway was investigated in the present study. AML12 cells were
treated with a range of concentrations of XML (0-4 mg/ml), and no
significant differences in cell viability were observed (Fig. 2A). Furthermore, the viability of
AML12 cells induced by LPS was detected via the MTT assay. The
viability of LPS-induced AML12 cells was significantly decreased
compared with the untreated group; however, cell viability
increased following treatment with XML in a concentration-dependent
manner; furthermore, the results showed a significant increase in
cell viability compared with the LPS group following 2 and 4 mg/ml
XML treatment (Fig. 2B).
Therefore, 4 mg/ml XML was used for subsequent experiments. The
expression levels of the proteins in the AMPK/PGC-1α signaling
pathway were detected using western blot analysis. The expression
levels of p-AMPK and PGC-1α were significantly decreased following
LPS stimulation compared with the control group, the effects of
which were reversed following 4 mg/ml XML intervention (Fig. 2C), suggesting that XML may activate
the AMPK/PGC-1α signaling pathway in LPS-induced AML12 cells.
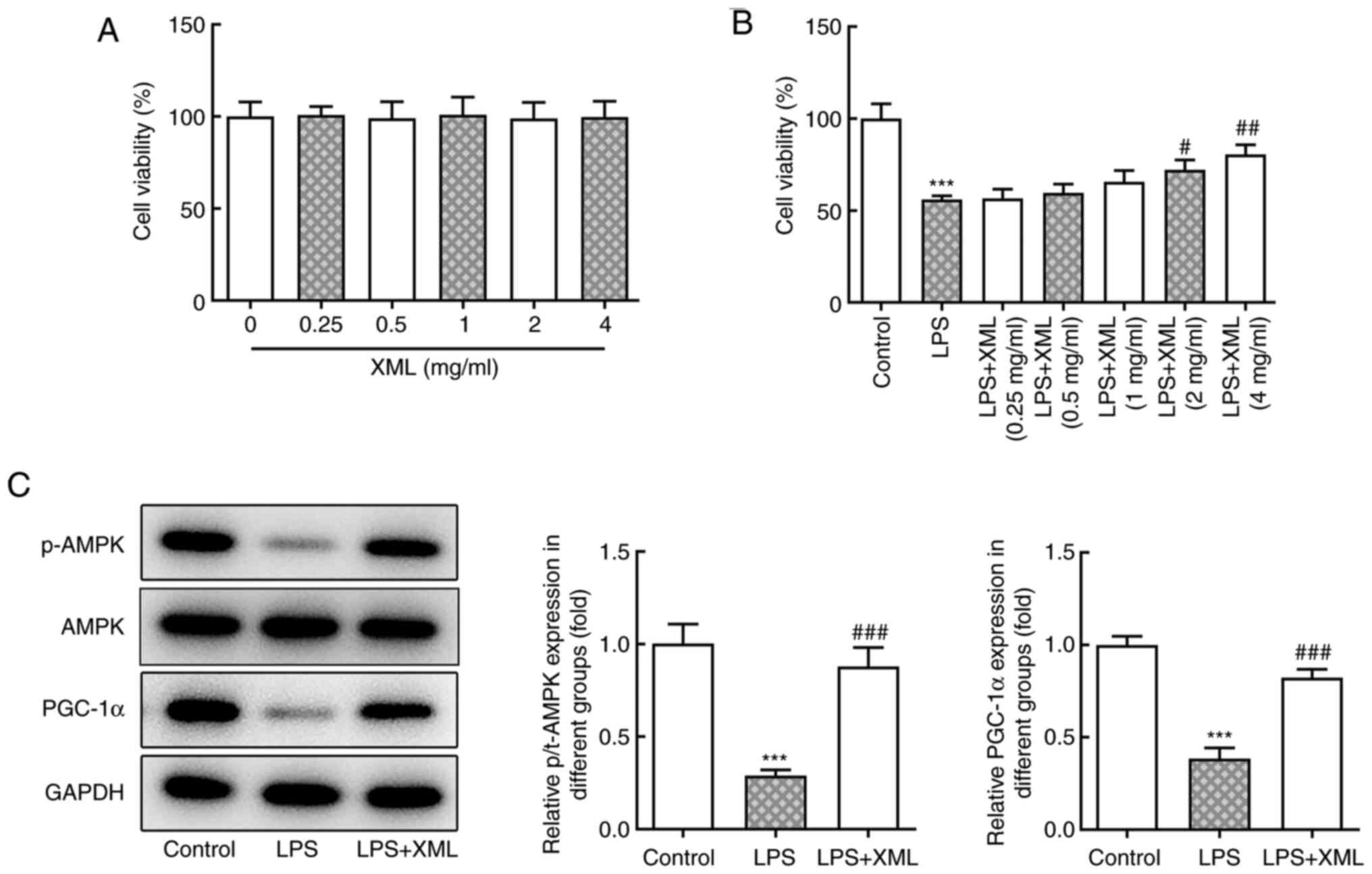 | Figure 2XML treatment activates the
AMPK/PGC-1α signaling pathway in LPS-stimulated AML12 cells. (A)
MTT assay was used to detect the viability of AML12 cells treated
with different concentrations of XML (0.25, 0.5, 1, 2 and 4 mg/ml).
(B) MTT assay was used to examine the effect of different
concentrations of XML (0.25, 0.5, 1, 2 and 4 mg/ml) on the
viability of 100 ng/ml LPS-induced AML12 cells. (C) Western blot
analysis was performed to detect the expression levels of proteins
in the AMPK/PGC-1α signaling pathway in 100 ng/ml LPS-induced AML12
cells after 4 mg/ml XML treatment. ***P<0.001 vs.
control; #P<0.01, ##P<0.01,
###P<0.001 vs. LPS. XML, xinmailong; AMPK,
AMP-activated protein kinase; PGC-1α, proliferator-activated
receptor γ coactivator-1α; LPS, lipopolysaccharide; p,
phosphorylated; t, total. |
XML treatment suppresses LPS-induced
inflammation, oxidative stress and apoptosis by activating
AMPK/PGC-1α signaling in AML12 cells
To determine the potential mechanism via which XML
exerts its effects on LPS-induced AML12 cell injury, the AMPK
inhibitor compound C (2 mg/ml) was used for pretreatment together
with 4 mg/ml XML in LPS-induced AML12 cells. The levels of
inflammatory factors were detected using ELISA and RT-qPCR
analysis. As presented in Fig.
3A-F, LPS significantly increased the levels of inflammatory
factors compared with the control group, including TNF-α, IL-6 and
IL-1β, and these levels were significantly decreased following
treatment with XML. Notably, combined treatment with XML and
compound C increased the expression levels of these factors
compared with the LPS+XML group (Fig.
3A-F). In addition, LPS induction significantly elevated the
content of MDA, accompanied by significantly decreased SOD
activity, compared with the group without LPS; these effects were
attenuated following treatment with XML compared with the LPS
treatment group. Furthermore, compound C intervention reversed the
effect of XML on oxidative stress (Fig. 3G and H). The results of the TUNEL assay further
demonstrated the ability of XML to inhibit the apoptosis of
LPS-induced AML12 cells, as the apoptosis of AML12 cells triggered
by LPS decreased in the LPS+XML group compared with the LPS
intervention group (Fig. 4A and
B). However, the LPS+XML+compound
C group exhibited a higher apoptotic rate compared with the LPS+XML
group. The expression of apoptosis-related proteins determined via
western blot analysis exhibited a similar trend (Fig. 4C). Collectively, these results
suggested that XML may suppress LPS-induced liver injury by
activating the AMPK/PGC-1α signaling pathway.
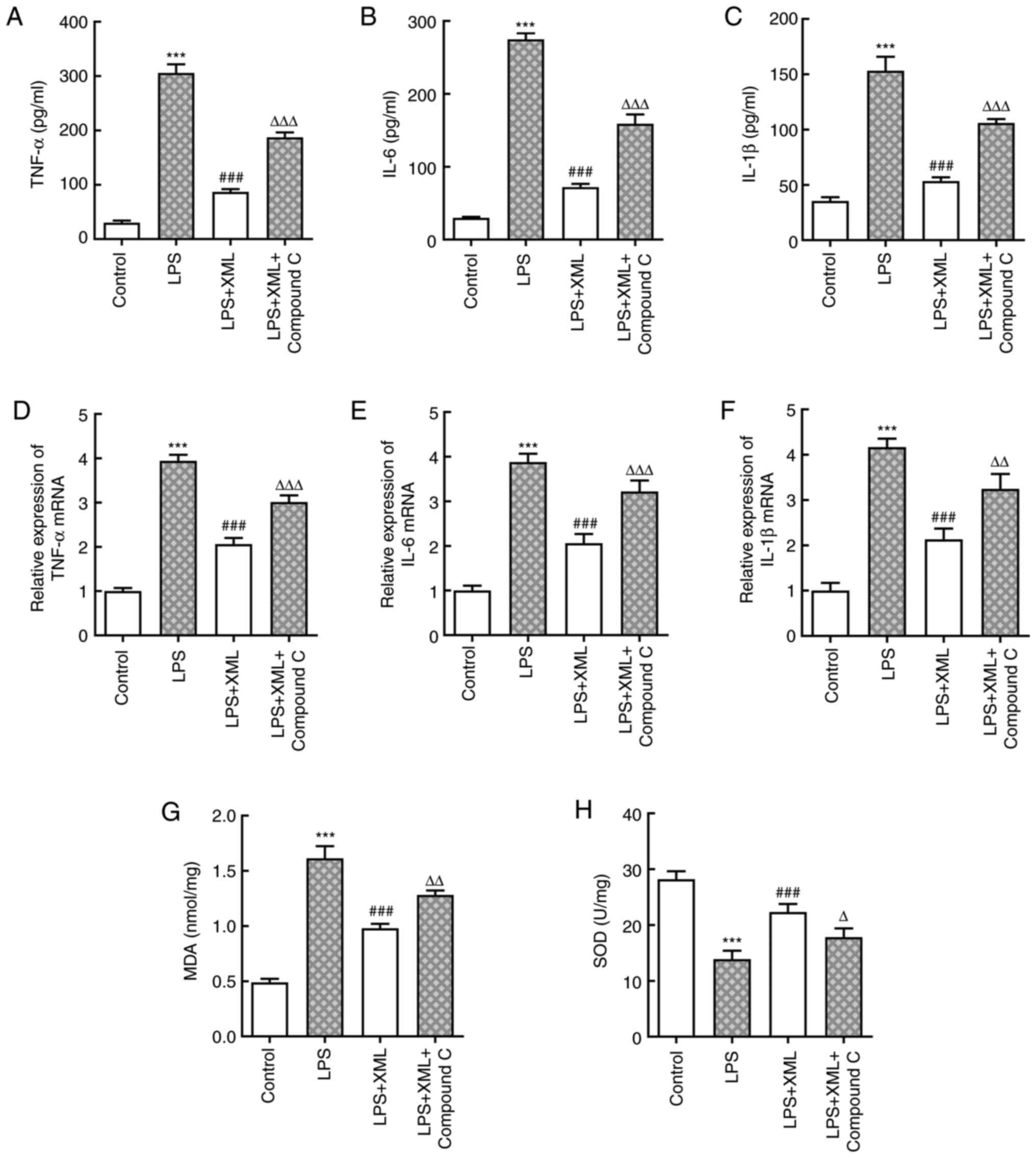 | Figure 3XML treatment suppresses LPS-induced
inflammation and oxidative stress in AML12 cells via the activation
of AMP-activated protein kinase/proliferator-activated receptor γ
coactivator-1α signaling. Levels of (A) TNF-α, (B) IL-6 and (C)
IL-β were detected using commercial ELISA kits. mRNA expression
levels of (D) TNF-α, (E) IL-6 and (F) IL-β were determined using
reverse transcription-quantitative PCR. Levels of (G) MDA and (H)
SOD were evaluated using corresponding kits. LPS, 100 ng/ml; XML, 4
mg/ml; compound C, 2 mg/ml. ***P<0.001 vs. control;
###P<0.001 vs. LPS; ∆P<0.05,
∆∆P<0.01, ∆∆∆P<0.001 vs. LPS+XML. XML,
xinmailong; LPS, lipopolysaccharide; MDA, malondialdehyde; SOD,
superoxide dismutase. |
XML suppresses mitochondrial
dysfunction induced by LPS by activating AMPK/PGC-1α signaling in
AML12 cells
Liver dysfunction is manifested by mitochondrial
dysfunction, and mitochondrial dysfunction and cellular energy
depletion enhance acute liver injury induced by sepsis (18,19).
Thus, it was hypothesized that XML may suppress mitochondrial
dysfunction induced by LPS by activating the AMPK/PGC-1α signaling
pathway, thereby resulting in the alleviation of hepatocellular
injury induced by LPS. LPS significantly decreased the content of
mitochondrial cytochrome c and significantly increased the
content of cytosolic cytochrome c compared with the
untreated group, and these effects were reversed following
treatment with XML (Fig. 5A and
B). However, the addition of
compound C significantly increased cytosolic cytochrome c
content compared with the LPS+XML group and decreased in
mitochondrial cytochrome c content, although this was not
statistically significant (Fig. 5A
and B). The synthesis of ATP,
reduced by LPS, was significantly enhanced following treatment with
XML compared with the LPS group, which was reversed by compound C
(Fig. 5C). The mRNA expression
levels of NRF1 and TFAM were detected via RT-qPCR analysis. As
presented in Fig. 5D and E, LPS significantly downregulated the
expression levels of NRF1 and TFAM, which was abrogated following
treatment with XML. Furthermore, compound C markedly downregulated
the expression levels of NRF1 and TFAM compared with the LPS+XML
group. Collectively, these results indicated that XML may suppress
mitochondrial dysfunction induced by LPS by activating the
AMPK/PGC-1α signaling pathway.
 | Figure 5XML suppresses the mitochondrial
dysfunction induced by LPS via the activation of AMP-activated
protein kinase/proliferator-activated receptor γ coactivator-1α
signaling in AML12 cells. Contents of (A) mitochondrial and (B)
cytosolic cytochrome c were measured using corresponding
kits. (C) Synthesis of ATP was evaluated using a
luciferin-luciferase reaction. The relative mRNA levels of (D) NRF1
and (E) TFAM were detected using reverse transcription-quantitative
PCR. LPS, 100 ng/ml; XML, 4 mg/ml; compound C, 2 mg/ml.
***P<0.001 vs. control; ##P<0.01,
###P<0.001 vs. LPS; ∆P<0.05,
∆∆P<0.01, ∆∆∆P<0.001 vs. LPS+XML. XML,
xinmailong; LPS, lipopolysaccharide; NRF1, nuclear respiratory
factor 1; TFAM, transcription factor A, mitochondrial. |
Discussion
PA extract is reported to exert therapeutic effects
in various diseases, such as fever, inflammation and chronic heart
failure (20). Furthermore, the
bioactive compounds of PA extract exert anti-inflammatory,
antipyretic, antioxidant and analgesic effects by suppressing the
MAPK/NF-κB signaling pathway (20). Previous reports have also indicated
the significance of PA extract in immune enhancement, wound
healing, anti-fibrosis and protection against viral infections
(12,13,21).
Additionally, PA extract attenuated gastrointestinal complications,
and improved immune function and nutritional status in patients
with systemic inflammatory response syndrome (12). Of note, PA extract accelerated
liver regeneration in a mouse liver regeneration model following
70% partial hepatectomy and a hepatocyte culture via a complex
network of pathways, including the AMPK signaling pathway (14). However, the beneficial effects and
mechanisms underlying PA extract in sepsis-induced hepatocyte
injury remain to be elucidated. The present study utilized XML and
established an LPS-induced liver injury model in vitro to
determine the underlying molecular mechanism. The results
demonstrated that XML ameliorated LPS-induced hepatocyte damage by
improving mitochondrial dysfunction via the AMPK/PGC-1α signaling
pathway.
Mitochondrial dysfunction leads to an impairment of
fat homeostasis and overproduction of reactive oxygen species in
the liver, which play important roles in the promotion of liver
injury (22,23). LPS induces ATP deficiency in
mitochondria, resulting in cellular dysfunction and cell death
(24). The promoter of cytochrome
c comprises a recognition site for the transcription factor
NRF1, which is responsible for the regulation of the expression of
genes involved in the respiratory chain. Furthermore, NRF1 has the
ability to activate TFAM (25,26).
In the present study, the translocation of mitochondrial cytochrome
c to the cytoplasm, a decrease in ATP levels and decreased
mitochondrial gene expression demonstrated the presence of
mitochondrial dysfunction in AML12 cells.
The degradative products of cells are eliminated by
a process referred to as autophagy (27). Autophagy maintains cellular
homeostasis, while imbalance in the level of autophagy is
associated with tissue injury, cancer and liver-related diseases
(28). Mitophagy, a specific type
of autophagy that removes damaged mitochondria, restrains
mitochondrial dysfunction triggered by oxidative stress and
decreases the accumulation of mutations in mitochondrial DNA
(29). Defects in mitophagy incur
mitochondrial dysfunction under conditions of liver injury induced
by ischemic reperfusion (30). The
regulation of autophagy is a complex process involving various
signaling pathways, including the AMPK-mediated signaling pathway
(31,32). AMPK indirectly facilitates
mitophagy and cell survival (33).
In the present study, the role of XML in the AMPK/PGC-1α signaling
pathway was investigated.
AMPK is a key regulator of mitochondrial biogenesis,
and the downstream effector of AMPK, PGC-1α, is required to
increase the expression of genes associated with oxidative stress
following AMPK activation (34).
PGC-1α is a potent stimulator that promotes the biogenesis of
mitochondria and transcription of genes in the liver, heart and
skeletal muscle (35). Under
conditions such as fasting and physical exertion, where energy is
lacking or in demand, PGC-1α is induced or activated (36). In addition, the regulation of
PGC-1α by AMPK is crucial for the modulation of mitochondrial
biogenesis (37). In the present
study, the suppression of AML12 cell viability by LPS was
alleviated by increasing the concentration of XML. The expression
levels of p-AMPK and PGC-1α increased following addition of XML to
LPS-induced AML12 cells, suggesting that XML activates the
AMPK/PGC-1α signaling pathway. Thus, it was hypothesized that XML
exerts inhibitory effects on LPS-induced liver injury via the
AMPK/PGC-1α signaling pathway. The results of the functional
experiments confirmed that XML alleviated the effects on
LPS-induced liver injury, as inflammation- and oxidative
stress-related factors decreased following treatment of AML12 cells
with XML, the effects of which were reversed following treatment
with compound C. In addition, compound C promoted the apoptosis of
AML12 cells. There is a close association between mitochondrial
dysfunction and liver injury, and the results of the present study
demonstrated that XML suppressed mitochondrial dysfunction induced
by LPS by activating the AMPK/PGC-1α signaling pathway.
In conclusion, the results of the present study
demonstrated that XML suppressed mitochondrial dysfunction induced
by LPS by activating the AMPK/PGC-1α signaling pathway, which may
contribute to understanding of the underlying mechanism via which
XML alleviates liver injury.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by Traditional Chinese
Medicine Science and Technology Development Program of Jiangsu
Province (grant no. YB201991).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
WS and JL were responsible for conceiving and
designing the study. WS, LA, JZ and JL performed the experiments.
WS, LA and JL analyzed the data and reviewed the manuscript. WS, LA
and JZ were responsible for drafting the manuscript. JL interpreted
data and revised the final manuscript. All authors have read and
approved the final manuscript. WS, LA, JZ and JL confirm the
authenticity of all the raw data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Trefts E, Gannon M and Wasserman DH: The
liver. Curr Biol. 27:R1147–R1151. 2017.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Miyajima A, Tanaka M and Itoh T:
Stem/progenitor cells in liver development, homeostasis,
regeneration, and reprogramming. Cell Stem Cell. 14:561–574.
2014.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Strnad P, Tacke F, Koch A and Trautwein C:
Liver-guardian, modifier and target of sepsis. Nat Rev
Gastroenterol Hepatol. 14:55–66. 2017.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Angus DC, Linde-Zwirble WT, Lidicker J,
Clermont G, Carcillo J and Pinsky MR: Epidemiology of severe sepsis
in the United States: Analysis of incidence, outcome, and
associated costs of care. Crit Care Med. 29:1303–1310.
2001.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Singer M, Deutschman CS, Seymour CW,
Shankar-Hari M, Annane D, Bauer M, Bellomo R, Bernard GR, Chiche
JD, Coopersmith CM, et al: The third international consensus
definitions for sepsis and septic shock (sepsis-3). JAMA.
315:801–810. 2016.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Savio LEB, de Andrade Mello P, Figliuolo
VR, de Avelar Almeida TF, Santana PT, Oliveira SDS, Silva CLM,
Feldbrügge L, Csizmadia E, Minshall RD, et al: CD39 limits P2X7
receptor inflammatory signaling and attenuates sepsis-induced liver
injury. J Hepatol. 67:716–726. 2017.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Zhang H, Wei L, Zhang Z, Liu S, Zhao G,
Zhang J and Hu Y: Protective effect of Periplaneta americana
extract on intestinal mucosal barrier function in patients with
sepsis. J Tradit Chin Med. 33:70–73. 2013.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Zhao Y, Yang A, Tu P and Hu Z: Anti-tumor
effects of the American cockroach, Periplaneta americana.
Chin Med. 12(26)2017.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Li QJ, Wang ZG, Liu Q, Xie Y and Hu HL:
Research status of Periplaneta americana with analyses and
prospects of key issues. Zhongguo Zhong Yao Za Zhi. 43:1507–1516.
2018.PubMed/NCBI View Article : Google Scholar : (In Chinese).
|
|
10
|
Ma X, Hu Y, Li X, Zheng X, Wang Y, Zhang
J, Fu C and Geng F: Periplaneta americana ameliorates
dextran sulfate sodium-induced ulcerative colitis in rats by
Keap1/Nrf-2 activation, intestinal barrier function, and gut
microbiota regulation. Front Pharmacol. 9(944)2018.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Li J, Shi W, Zhang J and Ren L: To explore
the protective mechanism of PTEN-induced kinase 1 (PINK1)/parkin
mitophagy-mediated extract of Periplaneta americana on
lipopolysaccharide-induced cardiomyocyte injury. Med Sci Monit.
25:1383–1391. 2019.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Zhang HW, Wei LY, Zhao G, Yang YJ, Liu SZ,
Zhang ZY, Jing Z and Hu YL: Periplaneta americana extract
used in patients with systemic inflammatory response syndrome.
World J Emerg Med. 7:50–54. 2016.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Li D, Li W, Chen Y, Liu L, Ma D, Wang H,
Zhang L, Zhao S and Peng Q: Anti-fibrotic role and mechanism of
Periplaneta americana extracts in CCl4-induced hepatic
fibrosis in rats. Acta Biochim Biophys Sin (Shanghai). 50:491–498.
2018.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Zou Y, Zhang M, Zeng D, Ruan Y, Shen L, Mu
Z, Zou J, Xie C, Yang Z, Qian Z, et al: Periplaneta
americana extracts accelerate liver regeneration via a complex
network of pathways. Front Pharmacol. 11(1174)2020.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Xing W, Yang L, Peng Y, Wang Q, Gao M,
Yang M and Xiao X: Ginsenoside Rg3 attenuates sepsis-induced injury
and mitochondrial dysfunction in liver via AMPK-mediated autophagy
flux. Biosci Rep. 37(BSR20170934)2017.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Yang Q, Han B, Xue J, Lv Y, Li S, Liu Y,
Wu P, Wang X and Zhang Z: Hexavalent chromium induces mitochondrial
dynamics disorder in rat liver by inhibiting AMPK/PGC-1α signaling
pathway. Environ Pollut. 265(114855)2020.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Singer M: The role of mitochondrial
dysfunction in sepsis-induced multi-organ failure. Virulence.
5:66–72. 2014.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Yan J and Li S and Li S: The role of the
liver in sepsis. Int Rev Immunol. 33:498–510. 2014.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Nguyen T, Chen X, Chai J, Li R, Han X,
Chen X, Liu S, Chen M and Xu X: Antipyretic, anti-inflammatory and
analgesic activities of Periplaneta americana extract and
underlying mechanisms. Biomed Pharmacother.
123(109753)2020.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Alcantara-Neves NM, Veiga RV, Ponte JC, da
Cunha SS, Simões SM, Cruz ÁA, Yazdanbakhsh M, Matos SM, Silva TM,
Figueiredo CA, et al: Dissociation between skin test reactivity and
anti-aeroallergen IgE: Determinants among urban Brazilian children.
PLoS One. 12(e0174089)2017.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Chen P, Shen Y, Shi H, Ma X, Lin B, Xiao
T, Wu F, Zhu J, Li Z, Xiao J, et al: Gastroprotective effects of
kangfuxin-against ethanol-induced gastric ulcer via attenuating
oxidative stress and ER stress in mice. Chem Biol Interact.
260:75–83. 2016.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Song Q, Xie Y, Gou Q, Guo X, Yao Q and Gou
X: JAK/STAT3 and smad3 activities are required for the wound
healing properties of Periplaneta americana extracts. Int J
Mol Med. 40:465–473. 2017.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Paradies G, Paradies V, Ruggiero FM and
Petrosillo G: Oxidative stress, cardiolipin and mitochondrial
dysfunction in nonalcoholic fatty liver disease. World J
Gastroenterol. 20:14205–14218. 2014.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Kelly DP and Scarpulla RC: Transcriptional
regulatory circuits controlling mitochondrial biogenesis and
function. Genes Dev. 18:357–368. 2004.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Evans MJ and Scarpulla RC: Interaction of
nuclear factors with multiple sites in the somatic cytochrome
c promoter. Characterization of upstream NRF-1, ATF, and
intron Sp1 recognition sequences. J Biol Chem. 264:14361–14368.
1989.PubMed/NCBI
|
|
27
|
Levine B, Packer M and Codogno P:
Development of autophagy inducers in clinical medicine. J Clin
Invest. 125:14–24. 2015.PubMed/NCBI View
Article : Google Scholar
|
|
28
|
Doria A, Gatto M and Punzi L: Autophagy in
human health and disease. N Engl J Med. 368(1845)2013.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Pickles S, Vigié P and Youle RJ: Mitophagy
and quality control mechanisms in mitochondrial maintenance. Curr
Biol. 28:R170–R185. 2018.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Kim JS, Nitta T, Mohuczy D, O'Malley KA,
Moldawer LL, Dunn WA Jr and Behrns KE: Impaired autophagy: A
mechanism of mitochondrial dysfunction in anoxic rat hepatocytes.
Hepatology. 47:1725–1736. 2008.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Li Y and Chen Y: AMPK and autophagy. Adv
Exp Med Biol. 1206:85–108. 2019.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Glick D, Barth S and Macleod KF:
Autophagy: Cellular and molecular mechanisms. J Pathol. 221:3–12.
2010.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Egan DF, Shackelford DB, Mihaylova MM,
Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor
R, et al: Phosphorylation of ULK1 (hATG1) by AMP-activated protein
kinase connects energy sensing to mitophagy. Science. 331:456–461.
2011.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Bergeron R, Ren JM, Cadman KS, Moore IK,
Perret P, Pypaert M, Young LH, Semenkovich CF and Shulman GI:
Chronic activation of AMP kinase results in NRF-1 activation and
mitochondrial biogenesis. Am J Physiol Endocrinol Metab.
281:E1340–E1346. 2001.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Jäger S, Handschin C, St-Pierre J and
Spiegelman BM: AMP-activated protein kinase (AMPK) action in
skeletal muscle via direct phosphorylation of PGC-1alpha. Proc Natl
Acad Sci USA. 104:12017–12022. 2007.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Gerhart-Hines Z, Rodgers JT, Bare O, Lerin
C, Kim SH, Mostoslavsky R, Alt FW, Wu Z and Puigserver P: Metabolic
control of muscle mitochondrial function and fatty acid oxidation
through SIRT1/PGC-1alpha. EMBO J. 26:1913–1923. 2007.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Reznick RM, Zong H, Li J, Morino K, Moore
IK, Yu HJ, Liu ZX, Dong J, Mustard KJ, Hawley SA, et al:
Aging-associated reductions in AMP-activated protein kinase
activity and mitochondrial biogenesis. Cell Metab. 5:151–156.
2007.PubMed/NCBI View Article : Google Scholar
|
















