Introduction
Bladder outlet obstruction (BOO) is a common
urological disease that is mainly caused by benign prostate
hyperplasia, contracture of the bladder neck, stricture of the
urethra, deformity of the lower urinary tract and compression by
organs around the bladder (1).
Long-term BOO may result in bladder structure remodeling and
bladder detrusor muscle dysfunction, which may in turn lead to a
series of urinary tract symptoms, including frequent urination,
urination urgency and urinary incontinence, severely compromising
the quality of life of the patients (2). Currently, BOO affects ~1.1 billion
individuals worldwide, with the occurrence of BOO and associated
lower urinary tract symptoms increasing annually (3,4).
During the pathological process of BOO, smooth muscle hypertrophy
occurs in an attempt to overcome the increased urethral resistance,
which leads to the eventual decompensation to fibrosis (5). Bladder tissue fibrosis is a
pathological consequence that occurs in most, if not all, cases of
BOO. It has been previously reported that TGF-β1-induced porcine
bladder urothelial cells successfully simulated the process of
bladder fibrosis, emphasizing the critical role of TGF-β1 in the
induction of fibrosis (6). Further
reports revealed that TGF-β1 serves an important role in the
development of bladder fibrosis, and inhibition of TGF-β1-induced
bladder tissue fibrosis may prove to be a viable strategy for BOO
treatment (7,8).
Aquaporin (AQP)2 belongs to the AQP family of
proteins, which includes 13 subtypes (AQP0-12). Serving as
classical water channel proteins that are ubiquitously expressed in
mammalian tissues, AQPs act as hydrophobic and integral membrane
channel proteins that are involved in fluid transport (9,10).
AQP2 has been found to be expressed in various tissues, including
fallopian tubes, pancreatic islets, small intestine, kidney and
bladder (10). Kim et al
(11) previously investigated the
regulatory mechanism of AQPs in a BOO rat model, and found that the
expression of both AQP2 and AQP3 were elevated in the rat urinary
bladder, indicating that AQP2/3 may be involved in BOO. In
addition, AQP2 was mainly expressed in the epithelial cell layer
and detrusor smooth muscle (11).
However, the effects of AQP2 on the physiology of bladder
dysfunction remain unclear and require further investigation.
hTFtarget analysis (http://bioinfo.life.hust.edu.cn/hTFtarget#!/)
identified a predicted interaction between the Forkhead box O1
(FOXO1) and the AQP2 promoter. FOXO1 is a member of the FOXO
protein family that is known to be involved in various cellular
processes, including cell proliferation and differentiation
(12). The FOXO1 gene is located on
chromosome 13 and translates into FOXO1 protein. Substantial
evidence indicates that the function of FOXO1 depends on the
interaction with DNA, thereby modulating downstream targets
(13). It has been reported that
FOXO1 may serve as a tumor suppressor in urothelial cells to
prevent urothelial carcinogenesis and cancer growth (14). Additionally, FOXO1 was found to be
closely involved in fibrosis of multiple organs and has been
considered to be a promising target for anti-fibrosis therapy
(15). However, at present, the
role of FOXO1 in bladder fibrosis remains unclear, and the
potential effects of FOXO1 on BOO warrant further
investigation.
On the basis of the aforementioned findings, the
present study hypothesized that AQP2 may combine with FOXO1, which
may in turn be involved in the pathophysiology of bladder
dysfunction as a result of BOO. The aim of the present study was to
explore the role of AQP2 in TGF-β-induced urothelial cell fibrosis
and its potential mechanism of action.
Materials and methods
Cell culture and treatment
The SV-HUC-1 human urinary tract epithelial cell
line was obtained from American Type Culture Collection and
cultured in Ham's F12 medium (Sigma-Aldrich; Merck KGaA)
supplemented with 7% fetal bovine serum (HyClone; Cytiva) and 1%
penicillin/streptomycin. SV-HUC-1 cells were cultured in a
humidified incubator with 5% CO2 at 37˚C. SV-HUC-1 cells
were then stimulated with TGF-β1 (R&D Systems, Inc.) at 1, 5
and 10 ng/ml to mimic bladder fibrosis.
Reverse transcription-quantitative PCR
(RT-PCR) analysis
Total RNA was extracted from SV-HUC-1 cells using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.). In total, 1 µg RNA was reverse-transcribed into cDNA in
accordance with the protocols of PrimeScript™ RT reagent
kit (Takara Bio, Inc.). Subsequently, qPCR was performed using
SYBR® Premix EX Taq™ II (Takara Bio, Inc.).
The primers used were as follows: AQP2, forward,
5'-TGGGCCATATGTGCTATGGAGA-3' and reverse,
5'-AAGGACACTCAGGTGCCAGGA-3'; FOXO1, forward,
5'-GGGTTAGTGAGCAGGTTACAC-3' and reverse,
5'-TCCAATGGCACAGTCCTTATC-3'; and β-actin, forward,
5'-CATCCACGAAACTACCTTCAACTCC-3' and reverse,
5'-GAGCCGCCGATCCACACG-3'. For PCR, the following thermocycling
program was adopted: Initial denaturation at 94˚C for 5 min; 30
cycles of denaturation at 94˚C for 30 sec, annealing at 65˚C for 30
sec and extension at 72˚C for 30 sec and a final extension step at
72˚C for 10 min. The relative mRNA expression of AQP2 and FOXO1 was
calculated using the 2-ΔΔCq method (16), followed by normalization using
β-actin as the reference gene.
Western blotting
Total protein was extracted from SV-HUC-1 cells
using RIPA lysis buffer (Nanjing KeyGen Biotech Co., Ltd.). After
determining the protein concentration using a bicinchoninic acid
protein assay kit (Nanjing KeyGen Biotech Co., Ltd.), equal amounts
of protein (30 µg/lane) were separated by 12% SDS-PAGE and
transferred onto PVDF membranes. The membranes were blocked with 5%
skimmed milk at room temperature for 2 h, followed by incubation
with primary antibodies against AQP2 (1:500; cat. no. ab199975;
Abcam), E-cadherin (1:10,000; cat. no. ab40772; Abcam), N-cadherin
(1:1,000; cat. no. ab18203; Abcam), cytokeratin 5 (1:10,000; cat.
no. ab52635; Abcam), fibronectin (1:1,000; cat. no. ab2413; Abcam),
α-SMA (1:10,00; cat. no. ab5831; Abcam), FOXO1 (1:1,000; cat. no.
ab39670; Abcam) and GAPDH (1:1,000; cat. no. ab8245; Abcam), at 4˚C
overnight. On the following day, the membranes were washed with TBS
with 0.1% Tween-20 (TBST) for 5 min three times and incubated with
horseradish peroxidase-conjugated goat anti-rabbit secondary
antibodies (1:20,000; cat. no. ZB-2301; OriGene Technologies, Inc.)
at room temperature for 2 h. The blots were visualized using the
ECL chemiluminescent substrate (GE Healthcare).
Cell transfection
To overexpress or inhibit the expression of AQP2,
pcDNA 3.1-AQP2 and short hairpin (sh) RNA targeting AQP2
(shRNA-AQP2; cat. no. C01001) were obtained from Shanghai
GenePharma Co., Ltd. When the cell confluence reached 60-70%, cells
were transfected with pcDNA 3.1-AQP2 (15 nM), shRNA-AQP2 (500
ng/µl) or their corresponding negative controls (NC; pcDNA-NC (15
nM) and shRNA-NC (500 ng/µl) using Lipofectamine® 3000
(Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's instructions in a humidified incubator with 5%
CO2 at 37˚C for 48 h. Western blot analysis was
performed 48 h after transfection to examine transfection
efficacy.
Cell viability assay
Cell viability was examined using the Cell Counting
Kit-8 (CCK-8) assay (Dojindo Molecular Technologies, Inc.)
according to the manufacturer's instructions. In brief, the
transfected cells were incubated with 5 ng/ml TGF-β1 for 12, 24 and
36 h at 37˚C. A total of 10 µl CCK-8 solution were then added to
each well and the cells were incubated for another 3 h. The
absorbance of each well was then detected at 450 nm using a
microplate reader (Bio-Rad Laboratories, Inc.).
Cell migration assay
Cell migration ability was measured using the wound
healing assay. Briefly, the transfected cells were inoculated into
six-well plates (1x106 cells/well) for incubation. When
the cell confluence reached 100%, a linear scratch was created
using a 200-µl pipette tip. The cells were washed with PBS before
being incubated in FBS-free medium for 24 h. Images were captured
at 0 and 24 h using a light microscope (magnification, x100).
Chromatin immunoprecipitation (ChIP)
assay
ChIP assay was performed using the
SimpleChIP® Plus Sonication Chromatin IP kit (Cell
Signaling Technology, Inc.) according to the manufacturer's
instructions. After the indicated treatment, SV-HUC-1 cells were
fixed with 4% formaldehyde at room temperature for 10 min and
sonicated using a 10 sec on and 10 sec off mode for 12 cycles on
ice. An antibody against FOXO1 (cat. no. ab39670; Abcam) was used
for the immunoprecipitation experiments before the extent of AQP2
enrichment was analyzed by RT-qPCR. IgG (cat. no. ab90285; Abcam)
was used as an isotype control and input DNA was amplified for each
sample in parallel runs.
Luciferase reporter assay
A predicted interaction between FOXO1 and AQP2
promoter was found using the hTFtarget online tool (http://bioinfo.life.hust.edu.cn/hTFtarget#!/). The
pGL3 plasmid (Promega Corporation) containing the AQP2 promoter
region element was generated by site-directed mutagenesis and
subcloned into the pGL3-basic luciferase reporter vector. A mutant
type (MUT) and wild-type (WT) AQP2 promoter vector were produced by
GeneCopoeia Co., Ltd. (Guangzhou, China). These plasmids (500
ng/µl) were transfected into SV-HUC-1 cells alongside
pcDNA3.1-FOXO1 (15 nM) using Lipofectamine® 3000
(Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's instructions. The luciferase reporter activity was
detected using the Dual-luciferase reporter assay (Promega
Corporation) according to the manufacturer's instructions. The
luciferase intensities were determined by normalizing the Firefly
luminescence to Renilla luminescence.
Statistical analysis
Data are presented as the mean ± SD from at least
three independent experiments. Statistical analysis was performed
using SPSS software, version 20.0 (IBM Corp.). Comparisons among
different groups were analyzed by unpaired Student's t-test or
one-way ANOVA followed by Tukey's post hoc test. P<0.05 was
considered to indicate a statistically significant difference.
Results
AQP2 expression is downregulated in
TGF-β1-treated SV-HUC-1 cells
To establish the bladder fibrosis cell model in
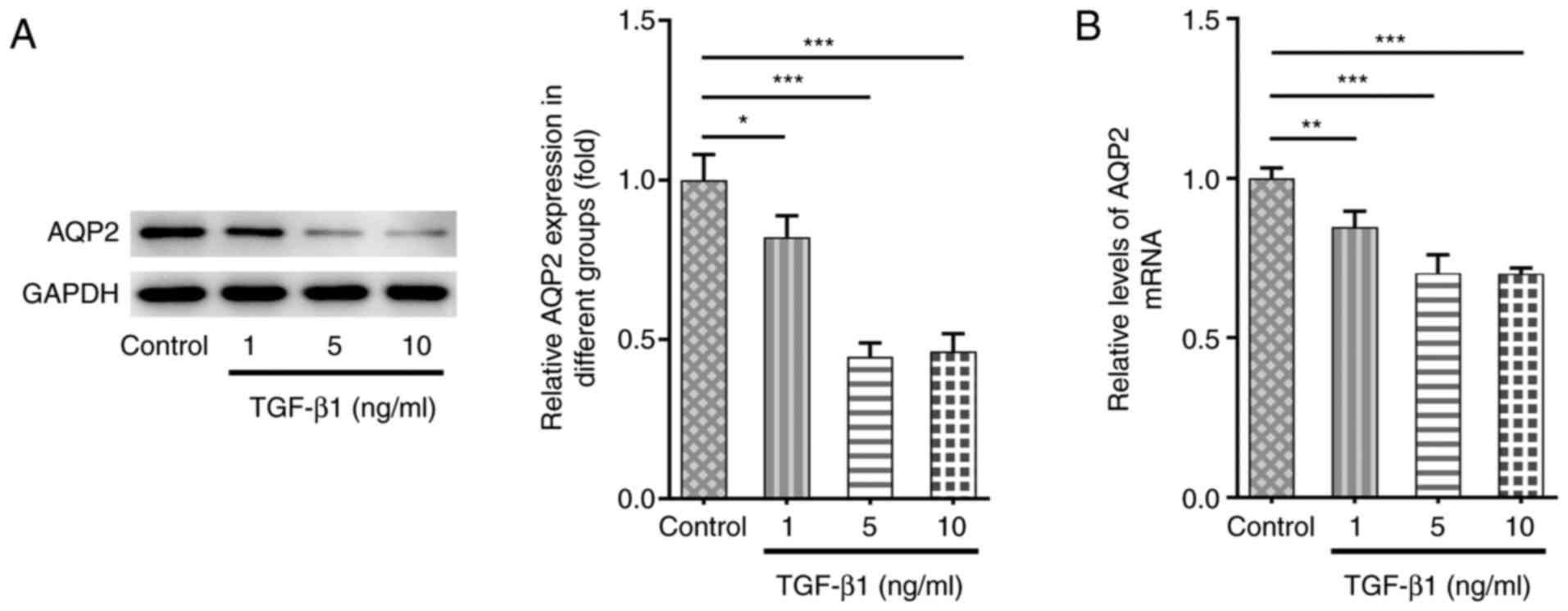
vitro, TGF-β1 was applied to stimulate SV-HUC-1 human urinary
tract epithelial cells. As shown in Fig. 1A and B, AQP2 expression was downregulated at
both the protein and mRNA levels following TGF-β1 stimulation
compared with untreated cells. Since there was no significant
difference in AQP2 expression between TGF-β1 treatment at 5 and 10
ng/ml, 5 ng/ml TGF-β1 treatment was selected for further
experiments.
AQP2 overexpression reduces cell
viability, inhibits migration and epithelial-to-mesenchymal
transition (EMT) in TGF-β1-induced SV-HUC-1 cells
To explore the role of AQP2 in bladder fibrosis,
SV-HUC-1 cells were transfected with pcDNA3.1-AQP2, which
significantly increased the protein expression of AQP2 (Fig. 2A). The cells were then treated with
TGF-β1, with or without AQP2 overexpression. TGF-β1 treatment
markedly increased cell viability, which was partially prevented by
AQP2 overexpression (Fig. 2B). The
wound healing assay revealed that TGF-β1 notably promoted cell
migration ability, which was also partially blocked by AQP2
overexpression (Fig. 2C and
D). Western blot analysis revealed
reduced expression of E-cadherin and increased expression of
N-cadherin after TGF-β1 treatment, suggesting that TGF-β1 promoted
EMT in SV-HUC-1 cells. Consistently, AQP2 overexpression also
partly prevented the effects of TGF-β1 on E-cadherin and N-cadherin
expression (Fig. 2E). TGF-β1
treatment was also found to induce bladder fibrosis, as the protein
expression level of cytokeratin was decreased, whereas the
expression of fibronectin (FN) and α-smooth muscle actin (α-SMA)
protein levels was increased following TGF-β1 treatment (Fig. 2F). These aforementioned changes were
also reversed by AQP2 overexpression.
 | Figure 2AQP2 overexpression reduces the
viability and inhibits migration and epithelial-to-mesenchymal
transition in TGF-β1-induced SV-HUC-1 cells. (A) SV-HUC-1 cells
were transfected with pcDNA3.1-NC or pcDNA3.1-AQP2, and the protein
expression of AQP2 was detected using western blotting.
***P<0.001 vs. pcDNA3.1-NC. (B-F) SV-HUC-1 cells were
transfected with pcDNA3.1-NC or pcDNA3.1-AQP2 and treated with
TGF-β1. (B) Subsequently, cell viability was measured using Cell
Counting Kit-8 assay. (C and D) Cell migration was measured using
wound healing assay. (E and F) E-cadherin, N-cadherin, cytokeratin,
FN and α-SMA protein expression levels were detected and analyzed
using western blotting. *P<0.05,
**P<0.01 and ***P<0.001 vs. control;
#P<0.05 and ###P<0.001 vs.
TGF-β1+pcDNA3.1-NC. AQP2, aquaporin 2; FN, fibronectin; AQP2,
aquaporin 2; α-SMA, α-smooth muscle actin; NC, negative
control. |
FOXO1 binds to the AQP2 promoter to
regulate AQP2 expression
Using the hTFtarget online tool (http://bioinfo.life.hust.edu.cn/hTFtarget#!/), a
predicted interaction between FOXO1 and AQP2 promoter was found
(Fig. 3A), which was verified using
luciferase reporter and ChIP assays (Fig. 3B and C). In addition, SV-HUC-1 cells were
transfected with pcDNA3.1-FOXO1 to overexpress FOXO1 (Fig. 3D). FOXO1 overexpression was found to
significantly increase both the protein and mRNA expression levels
of AQP2 in TGF-β1-induced SV-HUC-1 cells (Fig. 3E and F).
Inhibitory effects of FOXO1
overexpression on the viability, migration and fibrosis of
TGF-β1-induced SV-HUC-1 cells are partly abolished by AQP2
knockdown
In accordance with the interaction between FOXO1 and
AQP2, the expression level of FOXO1 in this in vitro model
of bladder fibrosis was also detected. The protein expression of
FOXO1 was found to be significantly decreased following TGF-β1
stimulation in SV-HUC-1 cells (Fig.
4A). shRNA-NC and shRNA-AQP-1/2 were subsequently used to
transfect SV-HUC-1 cells. AQP2 protein expression was found to be
significantly decreased in cells in the shRNA-AQP2-1 and
shRNA-AQP2-2 groups (Fig. 4B). Due
to its higher transfection efficacy, shRNA-AQP2-2 was selected for
further experiments. SV-HUC-1 cells were then transfected with
pcDNA-FOXO1, with or without shRNA-NC/shRNA-AQP2 co-transfection.
As shown in Fig. 4C-E, FOXO1
overexpression significantly reversed the increased cell viability
and migration abilities mediated by TGF-β1 in SV-HUC-1 cells. In
addition, FOXO1 overexpression increased the protein expression of
E-cadherin and cytokeratin, whilst reducing the protein expression
of N-cadherin, FN and α-SMA in TGF-β1-induced SV-HUC-1 cells
(Fig. 4F and G), suggesting that FOXO1 overexpression
also suppressed EMT and bladder fibrosis. However, the suppressive
effects of FOXO1 overexpression on cell viability, migration, EMT
and fibrosis were abolished by AQP2 knockdown (Fig. 4C-G).
 | Figure 4Inhibitory effects of FOXO1
overexpression on viability, migration and fibrosis in
TGF-β1-induced SV-HUC-1 cells are partly abolished by AQP2
knockdown. (A) FOXO1 protein expression was measured using western
blotting in SV-HUC-1 cells following TGF-β1 stimulation.
**P<0.01 vs. control. (B) shRNA-NC and shRNA-AQP-1/2
were used to transfect SV-HUC-1 cells and then AQP2 protein
expression was detected by western blotting. **P<0.01
and ***P<0.001 vs. control. (C) SV-HUC-1 cells were
treated with TGF-β1 and transfected with pcDNA-FOXO1 alone or
co-transfected with pcDNA-FOXO1 and shRNA-NC/shRNA-AQP2.
Subsequently, cell viability was measured using Cell Counting Kit-8
assay. (D and E) Cell migration was measured using wound healing
assay. (F and G) E-cadherin, N-cadherin, cytokeratin, FN and α-SMA
protein expression levels were detected and analyzed using western
blotting. *P<0.05 and ***P<0.001 vs.
control; #P<0.05, ##P<0.01 and
###P<0.001 vs. TGF-β1+Oe-NC; $P<0.05,
$$P<0.01 and $$$P<0.001 vs.
TGF-β1+Oe-FOXO1+shRNA-NC. FOXO1, forkhead box protein O1; AQP2,
aquaporin 2; shRNA, short hairpin RNA; NC, negative control; FN,
fibronectin; α-SMA, α-smooth muscle actin. |
Discussion
The aim of the present study was to elucidate the
role of AQP2 in TGF-β1-treated SV-HUC-1 cells and to investigate
the potential mechanism underlying the association between AQP2 and
bladder fibrosis. This was achieved by establishing an in
vitro cell model of bladder fibrosis using the SV-HUC-1 cell
line. A series of cellular function assays were performed, and the
data revealed that AQP2 expression was downregulated following
TGF-β1-induced fibrosis, whereas AQP2 overexpression could
alleviate the TGF-β1-mediated effects on cell viability, migration
and EMT. In addition, it was subsequently found that FOXO1 could
directly bind to the AQP2 promoter to regulate AQP2 expression,
where AQP2 upregulation downstream of FOXO1 inhibited the process
of bladder fibrosis.
EMT is a process during which differentiated
epithelial cells lose their epithelial phenotype and obtain
mesenchymal cell-like characteristics (11). Additionally, EMT has also been
previously associated with fibrosis (6). Mechanistically, loss of E-cadherin
expression is the initial and pivotal step in EMT (12,13).
However, the expression of other epithelial markers, such as
cytokeratin, is also lost (12,13).
This is accompanied by increases in the expression of mesenchymal
markers, including N-cadherin, FN and α-SMA (17,18).
Recently, TGF-β1 has been recognized as an inducer of EMT during
fibrotic events in several organs, including the lung, kidney and
liver (19-22).
However, corresponding data regarding bladder fibrosis remain
insufficient. To the best of our knowledge, only three previous
studies have reported that TGF-β1 may contribute to bladder
fibrosis by mediating EMT (6,18,23).
In the present study, TGF-β1 treatment resulted in the
downregulation of E-cadherin and cytokeratin expression, which was
accompanied by the upregulation of N-cadherin, FN and α-SMA
expression, revealing that TGF-β1 contributed to EMT in urinary
tract epithelial cells, consistent with the aforementioned previous
reports.
Binding of transcription factors to cognate DNA
sequences in promoter and enhancer regions of targeted genes is a
critical determinant of gene expression levels. FOXO1 is one of the
classical transcription factors that are regulated by cell surface
receptors. Promoter regions usually contain multiple transcription
factor-binding sites (24). A
present, only a small number of reports have revealed that FOXO1
can bind to the promoters of several genes, including STAT1 and
cyclin D1, to regulate multiple cellular processes, including cell
proliferation, migration and apoptosis (25,26).
In the present study, only one binding site for the transcription
factor FOXO1 was identified on the AQP2 promoter, which was
verified using luciferase reporter and ChIP assays, suggesting that
FOXO1 can interact with the DNA sequence in the promoter regions of
AQP2 to contribute to the upregulation of AQP2. Subsequent
experiments further revealed that FOXO1 overexpression upregulated
AQP2 expression, which participated in the regulation of bladder
fibrosis.
In conclusion, the results of the present study
demonstrated that the expression of AQP2 was downregulated in
TGF-β1-induced bladder fibrosis. FOXO1-induced upregulation of AQP2
expression was found to protect against TGF-β1-induced fibrosis by
reducing cell viability, migration and EMT. These results may
uncover a novel regulatory mechanism underlying bladder fibrosis
and indicate a novel biomarker target.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
ZL conceived and designed the study. YQ, ZX and ZG
performed the experiments, and performed data mining, acquisition
and analysis. ZL wrote and approved the final manuscript. ZL and YQ
confirmed the authenticity of all the raw data. All the authors
have read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
D'Silva KA, Dahm P and Wong CL: Does this
man with lower urinary tract symptoms have bladder outlet
obstruction? The rational clinical examination: A systematic
review. JAMA. 312:535–542. 2014.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Rademakers KL, van Koeveringe GA and Oelke
M: Detrusor underactivity in men with lower urinary tract
symptoms/benign prostatic obstruction: Characterization and
potential impact on indications for surgical treatment of the
prostate. Curr Opin Urol. 26:3–10. 2016.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Niemczyk G, Czarzasta K, Radziszewski P,
Wlodarski P and Cudnoch-Jedrzejewska A: Pathophysiological effect
of bladder outlet obstruction on the urothelium. Ultrastruct
Pathol. 42:317–322. 2018.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Wiafe B, Adesida AB, Churchill T, Kadam R,
Carleton J and Metcalfe PD: Mesenchymal stem cell therapy inhibited
inflammatory and profibrotic pathways induced by partial bladder
outlet obstruction and prevented high-pressure urine storage. J
Pediatr Urol. 15:254.e1–254.e10. 2019.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Metcalfe PD, Wang J, Jiao H, Huang Y, Hori
K, Moore RB and Tredget EE: Bladder outlet obstruction: Progression
from inflammation to fibrosis. BJU Int. 106:1686–1694.
2010.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Islam SS, Mokhtari RB, El Hout Y, Azadi
MA, Alauddin M, Yeger H and Farhat WA: TGF-β1 induces EMT
reprogramming of porcine bladder urothelial cells into collagen
producing fibroblasts-like cells in a Smad2/Smad3-dependent manner.
J Cell Commun Signal. 8:39–58. 2014.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Jiang X, Chen Y, Zhu H, Wang B, Qu P, Chen
R and Sun X: Sodium tanshinone IIA sulfonate ameliorates bladder
fibrosis in a rat model of partial bladder outlet obstruction by
inhibiting the TGF-β/Smad pathway activation. PLoS One.
10(e0129655)2015.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Chen Y, Ma Y, He Y, Xing D, Liu E, Yang X,
Zhu W, Wang Q and Wen JG: The TGF-β1 pathway is early involved in
neurogenic bladder fibrosis of juvenile rats. Pediatr Res: Jan 19,
2021 (Epub ahead of print).
|
|
9
|
Agre P and Kozono D: Aquaporin water
channels: Molecular mechanisms for human diseases. FEBS Lett.
555:72–78. 2003.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Niu D, Bai Y, Yao Q, Zhou L, Huang X and
Zhao C: AQP2 as a diagnostic immunohistochemical marker for
pheochromocytoma and/or paraganglioma. Gland Surg. 9:200–208.
2020.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Kim SO, Choi D, Song SH, Ahn KY, Kwon D,
Park K and Ryu SB: Effect of detrusor overactivity on the
expression of aquaporins and nitric oxide synthase in rat urinary
bladder following bladder outlet obstruction. Can Urol Assoc J.
7:E268–E274. 2013.PubMed/NCBI View
Article : Google Scholar
|
|
12
|
Wang Y, Zhou Y and Graves DT: FOXO
transcription factors: Their clinical significance and regulation.
Biomed Res Int. 2014(925350)2014.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Xing YQ, Li A, Yang Y, Li XX, Zhang LN and
Guo HC: The regulation of FOXO1 and its role in disease
progression. Life Sci. 193:124–131. 2018.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Ide H, Mizushima T, Jiang G, Goto T,
Nagata Y, Teramoto Y, Inoue S, Li Y, Kashiwagi E, Baras AS, et al:
FOXO1 as a tumor suppressor inactivated via AR/ERβ signals in
urothelial cells. Endocr Relat Cancer. 27:231–244. 2020.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Xin Z, Ma Z, Hu W, Jiang S, Yang Z, Li T,
Chen F, Jia G and Yang Y: FOXO1/3: Potential suppressors of
fibrosis. Ageing Res Rev. 41:42–52. 2018.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Polyak K and Weinberg RA: Transitions
between epithelial and mesenchymal states: Acquisition of malignant
and stem cell traits. Nat Rev Cancer. 9:265–273. 2009.PubMed/NCBI View
Article : Google Scholar
|
|
18
|
Wang J, Chen Y, Gu D, Zhang G, Chen J,
Zhao J and Wu P: Ketamine-induced bladder fibrosis involves
epithelial-to-mesenchymal transition mediated by transforming
growth factor-β1. Am J Physiol Renal Physiol. 313:F961–F972.
2017.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Lamouille S, Xu J and Derynck R: Molecular
mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell
Biol. 15:178–196. 2014.PubMed/NCBI View
Article : Google Scholar
|
|
20
|
Qian W, Cai X, Qian Q, Zhang W and Wang D:
Astragaloside IV modulates TGF-β1-dependent epithelial-mesenchymal
transition in bleomycin-induced pulmonary fibrosis. J Cell Mol Med.
22:4354–4365. 2018.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Iwano M: EMT and TGF-beta in renal
fibrosis. Front Biosci (Schol Ed). 2:229–238. 2010.PubMed/NCBI View
Article : Google Scholar
|
|
22
|
Shrestha N, Chand L, Han MK, Lee SO, Kim
CY and Jeong YJ: Glutamine inhibits CCl4 induced liver fibrosis in
mice and TGF-β1 mediated epithelial-mesenchymal transition in mouse
hepatocytes. Food Chem Toxicol. 93:129–137. 2016.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Lin S, Lian D, Liu W, Haig A, Lobb I,
Hijazi A, Razvi H, Burton J, Whiteman M and Sener A: Daily therapy
with a slow-releasing H2S donor GYY4137 enables early
functional recovery and ameliorates renal injury associated with
urinary obstruction. Nitric Oxide. 76:16–28. 2018.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Cardozo CP: Identification of
transcription factor-binding sites in the mouse FOXO1 promoter.
Methods Mol Biol. 1890:29–40. 2019.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Huang F, Wang Q, Guo F, Zhao Y, Ji L, An
T, Song Y, Liu Y, He Y and Qin G: FoxO1-mediated inhibition of
STAT1 alleviates tubulointerstitial fibrosis and tubule apoptosis
in diabetic kidney disease. EBioMedicine. 48:491–504.
2019.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Yang L, Yang F, Zhao H, Wang M and Zhang
Y: Circular RNA circCHFR facilitates the proliferation and
migration of vascular smooth muscle via miR-370/FOXO1/cyclin D1
pathway. Mol Ther Nucleic Acids. 16:434–441. 2019.PubMed/NCBI View Article : Google Scholar
|