Introduction
Renal fibrosis is recognized as the hallmark of the
majority of progressive renal diseases, including interstitial
fibrosis and glomerular sclerosis (1,2). Renal
interstitial fibrosis (RIF) is frequently the final outcome of
several chronic kidney diseases (CKDs), and is known as one of the
fundamental pathological changes leading to end-stage renal disease
(3-5).
Due to their high prevalence, CKDs and renal fibrosis affect ~10%
of the global population, particularly elderly individuals
(6,7). Since the majority of renal diseases
contribute to renal fibrosis, there is considerable interest in
investigating their underlying etiology to prevent or reverse the
aforementioned pathological changes.
The progression of CKD is characterized by the loss
of renal cells and their replacement by extracellular matrix (ECM).
A previous study demonstrated that the excessive deposition and
modification of the ECM in the renal parenchyma lead to
glomerulosclerosis and tubulointerstitial fibrosis (8). Unilateral ureteral ligation (UUO) is
one of the classical models of renal tubulointerstitial fibrosis,
which affects the collecting ducts and results in extensive
proximal tubular degeneration (9).
Moreover, growing evidence has indicated that the proximal tubular
injury caused by UUO is associated with RIF (10). Additionally, several molecules have
been found to be involved in the progression of RIF, including
TGF-β (3,11,12).
However, there are currently no effective treatment approaches for
preventing the onset and development of renal fibrosis. Therefore,
investigating the cellular and molecular mechanisms underlying RIF
progression is of great importance for its reversal or
elimination.
Hirudin, a thrombin inhibitor, is extracted from the
salivary gland of the medicinal leech Hirudo medicinalis
(13). Hirudin has gained
increasing interest as a potential treatment for renal fibrosis,
due to its relatively safe and inexpensive components that benefit
kidney health (14). Our previous
study showed that hirudin inhibited fibrosis in both renal tissues
and renal tubular epithelial cells via attenuation of inflammation,
regulating the expression of fibrosis- and epithelial-mesenchymal
transition (EMT)-related proteins, and reducing the apoptotic rate
of renal tubular epithelial cells (15). However, the molecular mechanism
underlying the effect of hirudin on RIF has not yet been
elucidated.
The non-hemostatic cellular effects of thrombin are
mediated by the activation of G-protein-coupled receptors, known as
protease-activated receptors (PARs). These receptors are expressed
on the surface of endothelial cells and numerous other cell types,
including proximal tubular epithelial cells (14). HK-2 cells express PAR1(16); therefore, the effect of hirudin on
renal fibrosis may be triggered by protease-activated receptor 1
(PAR1) signaling. Furthermore, another study demonstrated that
thrombin enhanced the activation of glomerular endothelial cells
via the sphingosine-1-phosphate (S1P)/sphingosine-1-phosphate
receptor 3 (S1PR3) signaling pathway (17), and a further study showed that S1P
upregulated the expression of PAR1 and PAR4(18). The aim of the present study was to
investigate the effects of hirudin on TGF-β-induced fibrosis in
renal proximal tubular epithelial cells, as well as its underlying
molecular mechanism.
Materials and methods
Animal model
All animal experiments were approved by the Guangxi
University of Chinese Medicine Institutional Animal Ethics and
Welfare Committee (approval no. DW20190107-013), and were carried
out according to the institutional guidelines for the care and use
of laboratory animals. In total, 60 male balb/c mice aged 12 weeks
(25±3 g weight) were obtained from the Laboratory Animal Center of
Peking University Health Science Center (license no.
DW20190107-013; Beijing, China). The animals were kept under
standard conditions at a room temperature of 25±2˚C with 60%
humidity with 12-h light-dark cycle and allowed free access to feed
and tap water. After 2 weeks of adaptive feeding, the mice were
randomly divided into four groups (n=15), including the control,
UUO, UUO + hirudin (10 mg/kg) and UUO + hirudin (15 mg/kg) groups.
The UUO mouse model was established as previously described
(12). Briefly, the mice were
anaesthetized with an intraperitoneal injection of 4% chloral
hydrate (370 mg/kg). No signs of peritonitis, pain or discomfort
were observed after anesthesia. Following the establishment of the
UUO model, two thirds of the UUO mice were administrated with 10 or
15 mg/kg hirudin on a daily basis. On day 21 after surgery, the
mice were sacrificed by cervical dislocation and the kidneys were
harvested.
Cell culture and treatment
Immortalized proximal tubular epithelial cells
(HK-2) were obtained from the American Type Culture Collection, and
maintained in 90% high-glucose DMEM (Thermo Fisher Scientific,
Inc.) supplemented with 10% FBS at 37˚C (5% CO2) in a
humidified atmosphere. Subsequently, the cells were cultured with
TGF-β (5 ng/ml; Sigma-Aldrich; Merck KGaA) for 48 h at 37˚C to
establish an in vitro RIF cell model. In addition, the HK-2
cells were treated with hirudin (0.5 or 1 mg/ml; Sigma-Aldrich;
Merck KGaA) in the presence or absence of 100 nM of S1P for 48 h at
37˚C or 50 µM of TFL (a PAR1 agonist; Sigma-Aldrich; Merck KGaA)
for 2 h at 37˚C .Cells were also treated with 5 ng/ml TGF-β,
followed by 10 µM JTE-013 (a S1PR2 antagonist; Sigma-Aldrich; Merck
KGaA) for 2 h at 37˚C or 1 µM TY52156 (a S1PR3 antagonist;
Sigma-Aldrich; Merck KGaA) for 2 h at 37˚C.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from HK-2 cells using a
TRIzol® reagent kit (Thermo Fisher Scientific, Inc.)
according to the manufacturer's instructions. RNA was reverse
transcribed into cDNA using the PrimeScript® RT reagent
kit (Takara Bio, Inc.) and the reaction was incubated at 25˚C for 5
min, 42˚C for 30 min, 85˚C for 5 min, and then kept at 4˚C for 5
min. Subsequently, qPCR was conducted using the SYBR Premix EX Taq™
II kit (Takara Bio, Inc.) on the PRISM 7500 system (Applied
Biosystems; Thermo Fisher Scientific, Inc.). The thermocycling
conditions were 95˚C for 3 min, followed by 35 cycles of
denaturation at 95˚C for 30 sec, annealing at 60˚C for 30 sec and
extension at 72˚C for 1 min. A final extension step at 72˚C for 7
min. The relative mRNA expression levels of the target genes were
determined using the 2-∆∆Cq method (19), and normalized to that of GAPDH. The
primer sequences were as follows: S1PR1 forward,
5'-TCTGCTCCTGCTTTCCA TCG-3' and reverse,
5'-AGGATGTCACAGGTCTTCGC-3'; S1PR2 forward,
5'-CCTTCGTGGCCAACACCTTA-3' and reverse, 5'-TGTCACTGCCGTAGAGCTTG-3';
S1PR3 forward, 5'-ACC GCGTGTTCCTTCTGATT-3' and reverse,
5'-TTGACCAGG CAGTAGATGCG-3'; S1PR4 forward, 5'-GTGTATGGCTG
CATCGGTCT-3' and reverse, 5'-CCACTAGGATGAGGGCG AAG-3'; PAR1
forward, 5'-TGTGAACTGATCATGTTTATG-3' and reverse,
5'-TTCGTAAGATAAGAGATATGT-3'; monocyte chemoattractant protein 1
(MCP-1), forward, 5'-GCTCAT AGCAGCCACCTTCATTC-3' and reverse,
5'-GTCTTCGGA GTTTGGGTTTGC-3'; and GAPDH forward, 5'-GGGAGC
CAAAAGGGTCATCATCT-3' and reverse, 5'-GACGCCTGC
TTCACCACCTTCTTG-3'.
Western blot analysis
The renal tissue samples and HK-2 cells were lysed
with RIPA buffer (Cell Signaling Technology, Inc.) supplemented
with protease inhibitors. A BCA Protein Assay kit (Beijing Dingguo
Changsheng Biotechnology Co., Ltd.) was used for protein
quantification, according to the manufacturer's instructions. A
total of 30 µg protein per lane was then separated by 10% SDS-PAGE
(Bio-Rad Laboratories, Inc.) and transferred onto a PVDF membrane
(EMD Millipore). Following blocking with 5% non-fat milk for 1 h at
room temperature, the membrane was incubated with specific primary
antibodies against S1PR1 (1:1,000; cat. no. ab242085; Abcam), S1PR2
(1:1,000; cat. no. ab235919; Abcam), S1PR3 (1:1,000; cat. no.
ab126622; Abcam), S1PR4 (1:1,000; cat. no. ab126392; Abcam), PAR1
(1:1,000; cat. no. 79109; Cell Signaling Technology, Inc.),
N-cadherin (1:1,000; cat. no. ab76011; Abcam), slug (1:1,000; cat.
no. ab27568; Abcam), E-cadherin (1:1,000; cat. no. ab231303;
Abcam), Collagen IV (1:1,000; cat. no. ab227616; Abcam),
fibronectin (FN; 1:1,000; cat. no. ab2413; Abcam), MMP9 (1:1,000;
cat. no. ab76003; Abcam), MCP-1 (1:500; cat. no. 81559; Cell
Signaling Technology, Inc.) and GAPDH (1:1,000; cat. no. 5174; Cell
Signaling Technology, Inc.) at 4˚C overnight. GAPDH was used as the
endogenous control. Following washing three times with TBS -0.5%
Tween-20, the membrane was incubated with the HRP-conjugated goat
anti-rabbit or mouse secondary antibodies (cat. nos. sc-2004 or
sc-2005; 1:5,000; Santa Cruz Biotechnology) for 1 h at room
temperature. The protein bands were visualized using ECL reagents
(NEN Life Science, Inc.) and analyzed by densitometry (QuantityOne
4.5.0 software; Bio-Rad Laboratories, Inc.).
Immunofluorescence analysis
HK-2 cells were seeded into 6-well plates at a
density of 5x104 cells/ml, and then treated with the
indicated compounds as aforementioned. The cells were washed with
PBS and then fixed with 4% paraformaldehyde for 30 min at room
temperature. Following washing with PBS, cells were blocked with 3%
BSA (Sigma-Aldrich; Merck KGaA) for 1 h at room temperature, and
then incubated with primary antibodies against α-smooth muscle
actin (α-SMA; dilution, 1:200; Abcam) at 4˚C overnight. The cells
were then incubated with Alexa Fluor 488-conjugated secondary
antibodies (dilution, 1:200; cat. no. A0428; Beyotime Institute of
Biotechnology) for 1 h at room temperature, washed with PBS and
stained with DAPI. Images were captured under an optical microscope
(CKX41; Olympus Corporation; magnification, x200).
Statistical analysis
The results are expressed as the mean ± SEM of at
least three independent experiments, and were analyzed using SPSS
18.0 statistical software (SPSS, Inc.). Statistical analysis was
performed using unpaired Student's t-test or one-way ANOVA followed
by Bonferroni's post hoc test. P<0.05 was considered to indicate
a statistically significant difference.
Results
S1PR1-4 and PAR1 are upregulated in
both TGF-β-induced HK-2 cells and renal tissues from mice with
UUO
To determine the pathological mechanism of RIF, the
expression levels of S1PR1-4 and PAR1 were evaluated both in
vivo and in vitro. RT-qPCR and western blot analyses
suggested that the relative mRNA and protein expression levels of
S1PR1-4 and PAR1 were increased in HK-2 cells in the TGF-β group,
compared with the control group (Fig.
1A and B). Additionally,
western blot analysis revealed that the protein expression levels
of these proteins were also significantly upregulated in renal
tissues from mice with UUO, compared with those in normal tissue
samples (Fig. 1C). Since both S1PR2
and S1PR3 were markedly upregulated, their roles were further
investigated in subsequent experiments.
Hirudin attenuates TGF-β-mediated
PAR1, S1PR2 and S1PR3 upregulation in HK-2 cells and renal tissues
from mice with UUO
To investigate the potential mechanism underlying
the effects of hirudin on RIF, the expression levels of PAR1, S1PR2
and S1PR3 were further assessed by RT-qPCR and western blot
analysis. As shown in Fig. 1D and
E, treatment of HK-2 cells with
hirudin decreased both the mRNA and protein expression levels of
PAR1 in a dose-dependent manner. Consistently, the protein
expression level of PAR1 was also downregulated in renal tissues
from mice with UUO following treatment with hirudin (Fig. 1F). Additionally, RT-qPCR and western
blotting demonstrated that treatment with increasing doses of
hirudin dose-dependently attenuated the expression of S1PR2 and
S1PR3 at both the mRNA (Fig. 2A)
and protein (Fig. 2B) levels in
TGF-β-induced HK-2 cells. Simultaneously, the protein expression
levels of S1PR2 and S1PR3 were significantly decreased by hirudin
treatment in the renal tissues of mice with UUO (Fig. 2C). The aforementioned findings
indicated that hirudin inhibited the TGF-β-mediated upregulation of
PAR1, S1PR2 and S1PR3 both in vivo and in vitro.
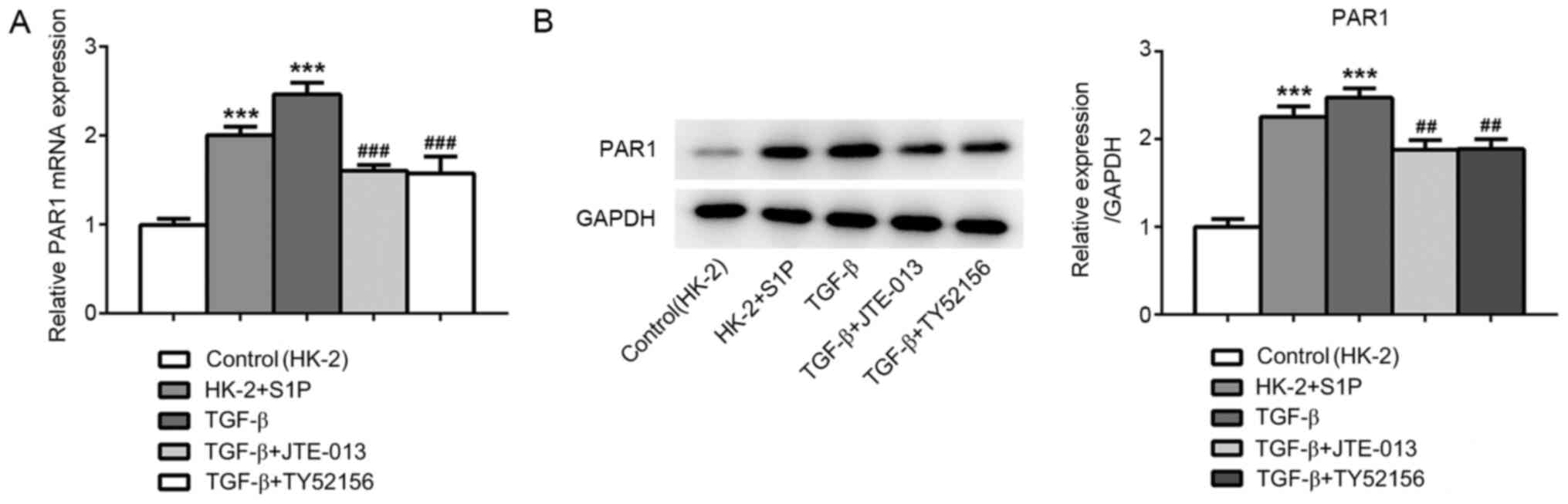
Inhibition of S1PR2 and S1PR3
suppresses PAR1 expression
To elucidate the mechanism underlying the effect of
hirudin on TGF-β-induced RIF, PAR1 expression in TGF-β-induced HK-2
cells was detected (20). RT-qPCR
and western blotting revealed that treatment with S1P and TGF-β
significantly upregulated PAR1, and the increased PAR1 level was
partially restored following treatment with JTE-013 or TY52156
(Fig. 3A and B). These results suggested that S1P could
promote PAR1 expression.
Hirudin attenuates TGF-β-induced EMT,
fibrosis and MCP-1 expression through S1P/S1PR2/S1PR3
signaling-mediated PAR1 downregulation
To further verify the molecular mechanism underlying
the effect of hirudin on TGF-β-induced fibrosis, HK-2 cells were
treated with PAR1 agonists (TFL; 50 µM) for 2 h. As shown in
Fig. 4A, western blotting revealed
that hirudin treatment significantly decreased the TGF-β-mediated
upregulation of N-cadherin and Slug, whilst restoring
TGF-β-mediated E-cadherin downregulation. Notably, exposure to S1P
and TFL reversed the effect of hirudin on the expression of
EMT-related proteins. The immunofluorescence assay results showed
that hirudin significantly attenuated TGF-β-induced α-SMA
upregulation, which was partially inhibited by S1P and TFL agonists
(Fig. 4B and C). Additionally, western blotting
demonstrated that hirudin significantly abrogated the
TGF-β-mediated upregulation of fibrosis-related proteins, including
collagen IV, FN and MMP9. Furthermore, exposure to S1P and TFL also
reversed the effect of hirudin on the expression of
fibrosis-related proteins (Fig.
5A). Finally, western blotting and RT-qPCR demonstrated that
hirudin treatment notably decreased TGF-β-induced MCP-1 protein and
mRNA expression levels, respectively. By contrast, treatment with
S1P and TFL restored the expression of MCP-1, even when combined
with hirudin (Fig. 5B and C), thus suggesting that hirudin attenuated
TGF-β-induced EMT, fibrosis and MCP-1 expression through PAR1
inhibition mediated via the S1P/S1PR2/S1PR3 signaling pathway.
Discussion
RIF is defined as a chronic and progressive process,
ultimately resulting in loss of renal function during aging and CKD
progression. Since RIF has a negative impact on patient quality of
life, it is necessary to determine effective therapeutic strategies
for RIF. Our previous study demonstrated that hirudin attenuated
fibrosis in renal tissues from mice with UUO, as well as inhibiting
inflammation, EMT and apoptosis in HK-2 renal tubular epithelial
cells (16). Therefore, the present
study aimed to further verify the effect of hirudin on RIF, and to
elucidate the potential mechanism underlying its effect.
Diabetes and hypertension are the two major risk
factors for CKD. It has been reported that the expression level of
the fibrogenic cytokine TGF-β is upregulated under hyperglycemic
conditions (21). Herein, HK-2
cells were stimulated with TGF-β to establish an in vitro
RIF cell model. The results demonstrated that the expression levels
of S1PR1-4 and PAR1 were significantly increased in both
TGF-β-induced HK-2 cells and renal tissues from mice with UUO,
particularly those of S1PR2 and S1PR3. Natural products have been
widely reported to protect against TGF-β-induced renal fibrosis
(22-24).
As one extract of natural products, hirudin abrogated TGF-β-induced
PAR1, S1PR2 and S1PR3 upregulation in both HK-2 cells and murine
renal tissues. A previous study demonstrated that the
TGF-β/sphingosine kinase 1/S1P signaling pathway promoted fibrosis
in renal tubular epithelial cells (25), which was consistent with the results
of the current study. Furthermore, the inactivation of S1P/S1PR2
signaling ameliorated diabetic nephropathy by restoring high
glucose-mediated intercellular adhesion molecule 1 and FN
upregulation in glomerular mesangial cells (26). Moreover, S1P upregulated the
expression of PAR1 and PAR4(18).
Herein, the inhibition of S1PR2 and S1PR3 markedly suppressed PAR1
expression. The aforementioned findings suggested that hirudin
could ameliorate TGF-β-induced RIF by PAR1 inhibition via the
S1P/S1PR2/S1PR3 signaling pathway. However, the specific
constituent of the S1P/S1PR2/S1PR3 pathway responsible for these
effects was not determined in the present study, and the regulatory
effects of S1P/S1PR2/S1PR3 signaling in renal fibrosis (and the
primary modulator therein) will be investigated in future
studies.
To further verify whether PAR1 and S1P/S1PR2/S1PR3
signaling could mediate the effect of hirudin on RIF development,
the expression levels of EMT- and fibrosis-related proteins were
determined by western blot analysis. EMT is characterized by
E-cadherin downregulation, and N-cadherin and Slug upregulation
(27). Additionally, α-SMA,
collagen-IV, FN and MMP9 are considered as reliable markers of EMT
in various organs during fibrosis (28). Therefore, EMT plays an essential
role in the pathogenesis of renal fibrosis. Herein, hirudin
significantly decreased the expression levels of N-cadherin, Slug,
α-SMA, collagen-IV, FN and MMP9, and increased that of E-cadherin.
Of note, S1P and PAR1 agonists abolished the effects of hirudin on
the expression of EMT- and fibrosis-related proteins.
Renal fibrosis is caused by the imbalance between
the excessive synthesis and reduced breakdown of ECM in the renal
parenchyma in response to injury and inflammation, eventually
resulting in the loss of renal function. The activation of PAR1 can
be induced by thrombin, which promotes proinflammatory responses in
endothelial cells (29). A previous
study showed that stimulation of renal tubular epithelial cells
with TGF-β promoted MCP-1 upregulation (30). Furthermore, PAR1 deletion resulted
in diminished production of MCP-1 in renal tissues from mice with
UUO (31). In the present study,
hirudin significantly downregulated MCP-1, which was reversed
following treatment with S1P and PAR1 agonists. Therefore, hirudin
may suppress TGF-β-induced EMT, fibrosis and MCP-1 expression via
the S1P/S1PR2/S1PR3 signaling-mediated inhibition of PAR1. Taken
together, the aforementioned results indicated that hirudin
attenuated TGF-β-induced fibrosis in renal proximal tubular
epithelial cells by downregulating PAR1 via S1P/S1PR2/S1PR3
signaling.
In summary, the current study revealed that hirudin
inhibited TGF-β-induced PAR1, S1PR2 and S1PR3 upregulation in both
HK-2 cells and renal tissues from mice with UUO. Additionally,
inhibition of S1PR2 and S1PR3 suppressed the expression of PAR1.
Collectively, hirudin attenuated TGF-β-induced EMT, fibrosis and
MCP-1 expression via PAR1 inhibition mediated by S1P/S1PR2/S1PR3
signaling, suggesting that hirudin administration may be a
promising effective strategy for RIF treatment.
Acknowledgements
Not applicable.
Funding
The present study was supported by National Natural Science
Foundation of China (grant no. 82060804), Guangxi University of
Chinese Medicine Science Research Program (grant no. 2020MS022),
the General Program of the Guangxi Natural Science Foundation
(grant no. 2020GXNSFAA259086) and the Guangxi Traditional Chinese
Medicine Appropriate Technology Development and Promotion Program
(grant no. GZSY20-27).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
QL and YX designed the experiments and wrote the
manuscript. QL, CL, ZW, RW and JM performed the experiments. WS and
JQ analyzed the data. JM searched the literature. All authors have
read and approved the final manuscript. YX and JM confirm the
authenticity of all the raw data.
Ethics approval and consent to
participate
All animal experiments were approved by the Guangxi
University of Chinese Medicine Institutional Animal Ethics and
Welfare Committee (approval no. DW20190107-013).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Martínez-Klimova E, Aparicio-Trejo OE,
Tapia E and Pedraza-Chaverri J: Unilateral ureteral obstruction as
a model to investigate fibrosis-attenuating treatments.
Biomolecules. 9(9)2019.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Wen Y, Rudemiller NP, Zhang J, Jeffs AD,
Griffiths R, Lu X, Ren J, Privratsky J and Crowley SD: Stimulating
type 1 angiotensin receptors on T lymphocytes attenuates renal
fibrosis. Am J Pathol. 189:981–988. 2019.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Li R, Guo Y, Zhang Y, Zhang X, Zhu L and
Yan T: Salidroside ameliorates renal interstitial fibrosis by
inhibiting the TLR4/NF-κB and MAPK signaling pathways. Int J Mol
Sci. 20(1103)2019.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Lv W, Fan F, Wang Y, Gonzalez-Fernandez E,
Wang C, Yang L, Booz GW and Roman RJ: Therapeutic potential of
microRNAs for the treatment of renal fibrosis and CKD. Physiol
Genomics. 50:20–34. 2018.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Nastase MV, Zeng-Brouwers J, Wygrecka M
and Schaefer L: Targeting renal fibrosis: Mechanisms and drug
delivery systems. Adv Drug Deliv Rev. 129:295–307. 2018.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Humphreys BD: Mechanisms of renal
fibrosis. Annu Rev Physiol. 80:309–326. 2018.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Chen Y, Mu L, Xing L, Li S and Fu S: Rhein
alleviates renal interstitial fibrosis by inhibiting tubular cell
apoptosis in rats. Biol Res. 52(50)2019.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Nogueira A, Pires MJ and Oliveira PA:
Pathophysiological mechanisms of renal fibrosis: A review of animal
models and therapeutic strategies. In Vivo. 31:1–22.
2017.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Ma Z, Wei Q, Zhang M, Chen JK and Dong Z:
Dicer deficiency in proximal tubules exacerbates renal injury and
tubulointerstitial fibrosis and upregulates Smad2/3. Am J Physiol
Renal Physiol. 315:F1822–F1832. 2018.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Forbes MS, Thornhill BA, Minor JJ, Gordon
KA, Galarreta CI and Chevalier RL: Fight-or-flight: Murine
unilateral ureteral obstruction causes extensive proximal tubular
degeneration, collecting duct dilatation, and minimal fibrosis. Am
J Physiol Renal Physiol. 303:F120–F129. 2012.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Isaka Y: Targeting TGF-β signaling in
kidney fibrosis. Int J Mol Sci. 19(19)2018.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Wang J, Zhu H, Huang L, Zhu X, Sha J, Li
G, Ma G, Zhang W, Gu M and Guo Y: Nrf2 signaling attenuates
epithelial-to-mesenchymal transition and renal interstitial
fibrosis via PI3K/Akt signaling pathways. Exp Mol Pathol.
111(104296)2019.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Wakui M, Fujimori Y, Nakamura S, Kondo Y,
Kuroda Y, Oka S, Nakagawa T, Katagiri H and Murata M: Distinct
features of bivalent direct thrombin inhibitors, hirudin and
bivalirudin, revealed by clot waveform analysis and enzyme kinetics
in coagulation assays. J Clin Pathol. 72:817–824. 2019.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Deng F, Zhang J, Li Y, Wang W, Hong D, Li
G and Feng J: Hirudin ameliorates immunoglobulin A nephropathy by
inhibition of fibrosis and inflammatory response. Ren Fail.
41:104–112. 2019.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Xie Y, Lan F, Zhao J and Shi W: Hirudin
improves renal interstitial fibrosis by reducing renal tubule
injury and inflammation in unilateral ureteral obstruction (UUO)
mice. Int Immunopharmacol. 81(106249)2020.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Bae JS, Kim IS and Rezaie AR: Thrombin
down-regulates the TGF-beta-mediated synthesis of collagen and
fibronectin by human proximal tubule epithelial cells through the
EPCR-dependent activation of PAR-1. J Cell Physiol. 225:233–239.
2010.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Sun XJ, Chen M and Zhao MH: Thrombin
contributes to anti-myeloperoxidase antibody positive IgG-mediated
glomerular endothelial cells activation through SphK1-S1P-S1PR3
signaling. Front Immunol. 10(237)2019.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Mahajan-Thakur S, Sostmann BD, Fender AC,
Behrendt D, Felix SB, Schrör K and Rauch BH:
Sphingosine-1-phosphate induces thrombin receptor PAR-4 expression
to enhance cell migration and COX-2 formation in human monocytes. J
Leukoc Biol. 96:611–618. 2014.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Wang GL, Xia XL, Li XL, He FH and Li JL:
Identification and expression analysis of the MSP130-related-2 gene
from Hyriopsis cumingii. Genet Mol Res. 14:4903–4913.
2015.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Hirata N, Yamada S, Shoda T, Kurihara M,
Sekino Y and Kanda Y: Sphingosine-1-phosphate promotes expansion of
cancer stem cells via S1PR3 by a ligand-independent Notch
activation. Nat Commun. 5(4806)2014.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Hsieh PF, Liu SF, Lee TC, Huang JS, Yin
LT, Chang WT, Chuang LY, Guh JY, Hung MY and Yang YL: The role of
IL-7 in renal proximal tubule epithelial cells fibrosis. Mol
Immunol. 50:74–82. 2012.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Chen HA, Chen CM, Guan SS, Chiang CK, Wu
CT and Liu SH: The antifibrotic and anti-inflammatory effects of
icariin on the kidney in a unilateral ureteral obstruction mouse
model. Phytomedicine. 59(152917)2019.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Chen DQ, Cao G, Zhao H, Chen L, Yang T,
Wang M, Vaziri ND, Guo Y and Zhao YY: Combined melatonin and
poricoic acid A inhibits renal fibrosis through modulating the
interaction of Smad3 and β-catenin pathway in AKI-to-CKD continuum.
Ther Adv Chronic Dis. 10(2040622319869116)2019.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Chen DQ, Feng YL, Cao G and Zhao YY:
Natural products as a source for antifibrosis therapy. Trends
Pharmacol Sci. 39:937–952. 2018.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Liu X, Hong Q, Wang Z, Yu Y, Zou X and Xu
L: Transforming growth factor-β-sphingosine kinase 1/S1P signaling
upregulates microRNA-21 to promote fibrosis in renal tubular
epithelial cells. Exp Biol Med (Maywood). 241:265–272.
2016.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Yang Z, Xiong F, Wang Y, Gong W, Huang J,
Chen C, Liu P and Huang H: TGR5 activation suppressed S1P/S1P2
signaling and resisted high glucose-induced fibrosis in glomerular
mesangial cells. Pharmacol Res. 111:226–236. 2016.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Man XY, Chen XB, Li W, Landeck L, Dou TT,
Chen JQ, Zhou J, Cai SQ and Zheng M: Analysis of
epithelial-mesenchymal transition markers in psoriatic epidermal
keratinocytes. Open Biol. 5(150032)2015.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Xu R, Chen MY, Liang W, Chen Y and Guo MY:
Zinc deficiency aggravation of ROS and inflammatory injury leading
to renal fibrosis in mice. Biol Trace Elem Res. 199:622–632.
2021.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Posma JJ, Grover SP, Hisada Y, Owens AP
III, Antoniak S, Spronk HM and Mackman N: Roles of coagulation
proteases and PARs (protease-activated receptors) in mouse models
of inflammatory diseases. Arterioscler Thromb Vasc Biol. 39:13–24.
2019.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Liang D, Song Z, Liang W, Li Y and Liu S:
Metformin inhibits TGF-beta 1-induced MCP-1 expression through
BAMBI-mediated suppression of MEK/ERK1/2 signalling. Nephrology
(Carlton). 24:481–488. 2019.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Waasdorp M, de Rooij DM, Florquin S,
Duitman J and Spek CA: Protease-activated receptor-1 contributes to
renal injury and interstitial fibrosis during chronic obstructive
nephropathy. J Cell Mol Med. 23:1268–1279. 2019.PubMed/NCBI View Article : Google Scholar
|