Introduction
Subarachnoid hemorrhage (SAH) is frequently observed
in severe diseases, and is associated with a high mortality ratio
and high disability rate worldwide (1). The mortality rate from SAH is ~50% in
population-based studies in the USA with a trend towards gradual
improvement (2,3). The majority of SAH-related studies
performed previously focused on cerebral vasospasm (CVS) following
SAH. Several treatments, such as circulatory volume expansion,
endothelin receptor antagonists and calcium antagonists, have been
applied to prevent vasospasm; however, the reversal of CVS did not
improve clinical outcomes following SAH (4).
Increasing numbers of clinical studies have reported
that the majority of post-SAH mortalities occur rapidly and are
caused largely by early brain injury (EBI), which begins to develop
within minutes after the initial bleeding started (5-7).
Mounting evidence shows that the alleviation of EBI increases
neurological function and ameliorates cognitive deficits in
experimental SAH (8,9). Therefore, EBI, which occurs within
24-72 h following SAH, may account for the abovementioned poor
outcomes following SAH.
The molecular mechanism underlying EBI development
after SAH is complex and involves cerebral edema, blood-brain
barrier (BBB) disruption or microvasculature dysfunction. Among the
mechanisms aforementioned, apoptosis has been revealed to be an
important player in EBI (10).
Endothelial apoptosis has been revealed to cause BBB breakdown and
consequently induce brain edema. Moreover, a study also revealed
that neuronal apoptosis is an essential part of EBI following SAH
(11), whereas inhibition of
apoptosis has been demonstrated to exert protective effects against
SAH, thereby decreasing mortality and improving neurological
outcomes in SAH animal models (12).
Cyclin-dependent kinase 5 (Cdk5), a proline-directed
serine-threonine kinase, is highly expressed in cells of the
nervous system (13). A previous
study has demonstrated that Cdk5 is involved in balancing survival
and apoptosis of neurons in the central nervous system (14). In contrast to canonical
Cdk-signaling, Cdk5 does not function in cell cycle regulation;
Cdk5 is instead involved in cytoskeletal protein phosphorylation,
neurogenesis, cognition and neuronal survival in the brain under
physiological conditions (15).
Cdk5 is also activated by the non-cyclin proteins p35 and p39,
which are predominantly expressed in the CNS under physiological
conditions (16). However, during
neurotoxic stress, Cdk5 binds with p25 (a Cdk5 activator), which
triggers Cdk5 hyperactivation. Aberrant Cdk5 activity leads to the
hyperphosphorylation of several downstream substrates, contributing
to cellular dysfunction and the development of neurological disease
(17).
Previous studies have indicated that expression
levels of Cdk5 and Cdk5 phosphorylated at Tyr15 (Cdk5-pTyr15) in
the human brain increased significantly after acute ischemic
stroke, and that inhibition of Cdk5 significantly reduced infarct
volumes on day 1 in animal stroke models (18-20).
However, the biological functions of Cdk5 in EBI after SAH are
still unclear. Thus, the present study aimed to investigate the
role of Cdk5 in EBI following SAH.
Materials and methods
Animal sourcing
In total, 138 male Sprague-Dawley rats (weight,
250-320 g; 6-8 weeks) were purchased from Jinling Hospital
(Nanjing, China). Rats were first acclimated to animal cabinets at
23±1˚C, with 100% fresh air, 50% humidity and free access to food
and water under 12-h light/dark cycles. All study protocols
regarding surgical procedures and animal usage were approved by The
Ethics Committee of the Affiliated Suqian Hospital of Xuzhou
Medical University (Suqian, China) and conformed to the Guide for
the Care and Use of Laboratory Animals (8th edition) by National
Research Council (US) Committee (21).
Induction of experimental SAH
The prechiasmatic injection model of SAH was
prepared by modifying some previous procedures (22-24).
Briefly, male Sprague-Dawley rats were anesthetized via
intraperitoneal injections of sodium pentobarbital (40 mg/kg). A
midline scalp incision was made and a 1-mm hole was created at a
location 7.5-8.0 mm above the bregma (25). A pre-chiasmatic cistern SAH
injection model (25) was
established by drawing 300 µl fresh autologous arterial blood from
the rat's femoral artery using insulin needles before drilling a
1-mm hole above the bregma. The blood was slowly injected into the
pre-chiasmatic cistern via the hole, which was fed into the hole
and then removed after blood was injected. After these steps, the
rats were returned to their home cages and fed normally. The room
temperature was set at 23±1˚C.
Experimental design
To analyze the expression levels of Cdk5,
Cdk5-pTyr15 and p25 after SAH, rats were randomly assigned into six
groups in time course experiments: Sham (n=6), 6 h after SAH (n=6),
12 h after SAH (n=6), 1 day after SAH (n=6), 3 days after SAH (n=6)
and 5 days after SAH (n=6). Immunofluorescence assay was used to
determine the cellular localization of Cdk5 (Sham, n=6; 1 day after
SAH, n=6). The rats in the sham group were injected with the same
volume of 0.9% saline into the bregma hole with reference to
procedures mentioned above.
To explore the role of Cdk5 following SAH, a total
of 90 rats were randomly assigned to four groups: i) A sham (20%
PBS) group (n=24); ii) a SAH + vehicle group (n=24); iii) a SAH +
roscovitine (50 µg) group (n=18); and iv) a SAH + roscovitine (100
µg) group (n=24). Roscovitine was administered at 30 min after
blood injection in the SAH + roscovitine groups. The same volume of
20% physiological salt solution was simultaneously administered to
the SAH + vehicle group. All rats were sacrificed on day 1 after
SAH after injection with intraperitoneal injection of 1% sodium
pentobarbital (150 mg/kg). Confirmation of rat death was confirmed
when the heart stopped beating. Next, the rats were transcardially
perfused with PBS via the left ventricle. After the blood clots
were cleared from the brain tissues, the temporal lobe was
dissected for further assays. The brain tissue samples (40 µm)
reserved for western blotting were immediately frozen in -80˚C
liquid nitrogen, whereas those for the TUNEL and Nissl staining
experiments were fixed with 10% neutral-buffered formalin at 30˚C
for 24 h.
Drug administration
Roscovitine, a Cdk5 inhibitor purchased from Santa
Cruz Biotechnology, Inc., was dissolved in 20% dimethylsulfoxide
(physiological salt solution). The induction of anesthesia was
performed in a small induction chamber, where the flow of oxygen-4%
isoflurane is 2 l/min for 3-4.5 min. After 3 min, the foot was
gently pinched with tweezers at 30-sec intervals to test the
anesthesia state. Roscovitine (50 or 100 µg in 10 µl of 20%
physiological salt solution) or the same volume of physiological
salt solution was intracerebroventricularly administered into each
rat at 30 min after sham injury or SAH during, which a new 1.0-mm
lateral hole at 1.5 mm behind the bregma was made, through which
the drug was administered. The drug dose utilized in this study was
selected based on a previous study using a traumatic brain injury
model (26).
Western blotting
Brain tissues isolated from the temporal cortex were
homogenized. The samples were subsequently diluted (1:1) in RIPA
buffer (50 mM Tris-HCl pH 7.2, 150 mM NaCl, 1% NP-40, 0.1% SDS,
0.5% DOC, 1 mM PMSF, 25 mM MgCl2 and supplemented with a
phosphatase inhibitor cocktail) and boiled for ≥5 min at 100˚C.
Protein concentration was estimated using the Bradford method
(27). In total, 20 µg proteins
were then separated by 10% SDS-PAGE and transferred onto a
polyvinylidene fluoride membrane, which was blocked for 30 min in
5% non-fat milk in 1X TBS-0.1% Tween at 37˚C and then incubated
with primary antibodies at 37˚C for Cdk5 (1:1,000, cat. no.
ab40773; Abcam), Cdk5-pTyr15 (1:1,000; cat. no. AP55874PU-N;
OriGene Technologies, Inc.), p25 (1:1,000; cat. no. ab125653;
Abcam) and β-actin (1:1,000; cat. no. ab8226; Abcam) overnight. The
membranes were then incubated with secondary antibodies [1:1,000;
goat anti-rabbit IgG H&L (HRP); cat. no. ab97051; Abcam] for 1
h at 37˚C and bands of blotted protein were visualized using
enhanced chemiluminescence (Thermo Fisher Scientific, Inc.). All
data was normalized to the corresponding expression level of
β-actin using ImageJ 1.8.0 (National Institutes of Health).
Immunofluorescence staining
Double immunofluorescence staining experiments were
performed as per methods previously described by our laboratory
(22). Before immunofluorescence
staining, frozen temporal cortex tissue sections (6-µm) were warmed
at 26˚C for 30 min and fixed in ice-cold acetone or other alternate
4% paraformaldehyde fixatives for 10 min. The sections were blocked
in 5% FBS for 60 min and incubated with primary antibodies
(Anti-NeuN antibody-Neuronal Marker; 1:100; cat. no. ab177487;
Abcam; Anti-GFAP antibody; 1:100; cat. no. ab7260; Abcam; and
Cdk5-antibody; 1:50; cat no sc-6247; Santa Cruz Biotechnology,
Inc.) for 1 h at room temperature. The sections were washed and
incubated with appropriate secondary antibodies (Alexa
Fluor® 594-conjugated goat anti-rabbit IgG H&L;
1:300, cat. no. ab150080; Abcam; and Alexa Fluor®
488-conjugated goat anti-mouse IgG H&L; 1:300, cat. no.
ab150113; Abcam) for 1 h at 26˚C. Cell nuclei were stained using
4-diamidino-2-phenylindole (DAPI). Fluorescence microscopy was
performed with a ZEISS HB050 inverted microscope (magnification,
x40; Carl Zeiss AG) and six views of fields were processed using
Image-Pro Plus 7.0 (Media Cybernetics, Inc.).
Brain water content
After the rats were sacrificed, the cerebella were
removed and brain hemispheres were dissected to determine the wet
weights. Brain water content was determined using the following
formula [(wet weight-dry weight)/wet weight] x100%. The brains were
fixed using 4% paraformaldehyde for 30 min at room temperature.
Nissl staining
The same portion of the temporal cortex was used
from each rat for Nissl staining. After deparaffinization and
rehydration, the slice was wash in 100% ethanol twice for 10 min
each, then twice 95% in ethanol for 10 min each. It was then washed
in deionized H2O for 1 min with stirring. The slides
were placed in a container and covered with 10 mM sodium citrate
buffer, pH 6.0; or with 50 mM glycine-HCl buffer (glycine:
sc-29096), pH 3.5, with 0.01% (w/v) EDTA (EDTA: sc-29092) and
heated at 95˚C for 10 min. The 6-µm sections were stained with 1%
toluidine blue for 2-3 min at room temperature. The slices were
rinsed with distilled water, and washed with 70, 95 and 100%
ethanol for differentiation. Subsequently, the sections were
dehydrated in an ascending xylene series and mounted using a light
microscope (Leica DM750M; Leica Microsystems GmbH (x400
magnification) in six randomly selected sections were subsequently
evaluated using a light microscope and surviving neurons in each
section were averaged. The average number of surviving neurons
across three sections was considered the number of surviving
neurons for each sample. All analyses were performed by three
pathologists with no knowledge of group assignments.
TUNEL staining
Apoptotic cells were detected using a TUNEL
detection kit (QIA 33; Merck & Co., Inc.) using the protocol
previously described (28).
Briefly, after washing with PBS, the proteinase K (20 g/ml,
Sigma-Aldrich; Merck KGaA)-digested tissues were incubated in 3%
hydrogen peroxid-PBS, following which they were incubated with
TUNEL reaction fluid (Roche Diagnostics GmbH) for 1 h at 37˚C. The
sections were then incubated with HRP-conjugated anti-digoxigenin
antibodies (1:10,000; cat. no. ab6212; Abcam) for 2 h at room
temperature. In total, 0.05% DAB was adopted to stain the sections
for 5-10 min at room temperature, which were lightly counterstained
with hematoxylin for 2 min at room temperature. Apoptotic cells
were analyzed in six fields of view (magnification, x40) using a
light microscope (Leica DM750M; Leica Microsystems GmbH) and were
processed using Image-Pro Plus 7.0 (Media Cybernetics, Inc.).
Neurological scores
Neurological scores were evaluated on day 1 after
SAH using the scoring method proposed by Garcia (29). The scoring system includes
categories for spontaneous activity (0-3), symmetry of movements
(0-3), symmetry of the forelimbs (0-3) and the climbing ability on
the cage wall (1-3), as well as sensory scores, which were
determined by assessing responses to touching of the vibrissae or
the sides of the trunk (1-3). All six individual scores were
summed, and the value obtained was considered the neurological
score for each rat.
Statistical analysis
Data are presented as means ± SEM and were analyzed
with SPSS 17.0 (SPSS, Inc.). All the data apart from neurological
score were compared using one-way ANOVA with post hoc Tukey's test.
Neurological score was analyzed using Kruskal-Wallis with post hoc
Dunn's Test. P<0.05 was considered to indicate a statistically
significant difference.
Results
Western blotting results of Cdk5,
Cdk5-pTyr15 and p25 expression
The sham group exhibited low expression levels of
Cdk5 and Cdk5-pTyr15 (Fig. 1A-C).
However, following SAH, Cdk5 and Cdk5-pTyr15 expression level were
significantly upregulated in the SAH group compared with those in
the sham group. The expression of Cdk5 peaked on day 1, whereas
Cdk5-pTyr15 expression peaked at 12 h following SAH (P<0.01;
Fig. 1B and C). The time course of p25 expression was
similar to those of Cdk5 and Cdk5-pTyr15, where p25 protein levels
increased significantly and peaked on day 1 after SAH (P<0.01;
Fig. 1D) in SAH group compared with
those in the sham group. The protein ratio of Cdk5-pTyr15/Cdk5
expression is presented in Fig. 1E.
Following SAH, the protein ratio of Cdk5-pTyr15/Cdk5 expression
were significantly regulated in the SAH group compared with those
in the sham group, which peaked at 12 h following SAH (P<0.01;
Fig. 1E).
Immunofluorescence staining of Cdk5
after SAH
Cdk5 was colocalized with neuronal nuclei in the
sham and SAH groups, suggesting that Cdk5 is expressed in both
healthy and post-SAH rat brain neurons. Following SAH, Cdk5
translocated to the nucleus in the SAH group but not in the sham
group (Fig. 2). Cdk5 was weakly
expressed in glial fibrillary acidic protein-positive cells of the
sham group; however, enhanced Cdk5 immunoreactivity was observed in
the astrocytes of the SAH group, demonstrating that Cdk5 is
activated in astrocytes following SAH (Fig. 2).
Roscovitine decreases Cdk5-pTyr15
expression
The SAH + roscovitine group exhibited significantly
upregulated Cdk5-pTyr15 expression in the temporal cortex on day 1
after SAH compared with the sham + vehicle group (P<0.01;
Fig. 3). Moreover, treatment with
roscovitine suppressed Cdk5-pTyr15 expression in a dose-dependent
manner in the SAH + roscovitine group, and treatment with high-dose
roscovitine (100 µg) significantly inhibited Cdk5-pTyr15 expression
in the SAH + roscovitine group compared with the SAH + vehicle
group (P<0.01; Fig. 3).
Influence of roscovitine on brain
edema and neurobehavioral deficits after SAH
SAH led to a significantly elevated level of brain
water content in the SAH + vehicle group compared with that in the
sham + vehicle group. Roscovitine (50 µg) administration failed to
alleviate brain edema; however, roscovitine (100 µg) administration
elicited a noticeable decrease in brain water content in the SAH +
roscovitine (100 µg) group in contrast to the SAH + vehicle group
(P<0.05; Fig. 4A). These
findings indicated that roscovitine attenuated brain edema.
The SAH + vehicle group demonstrated significantly
lower neurological scores compared with the sham + vehicle group;
however, the rats that received roscovitine (100 µg) after SAH
exhibited a significant increase in neurological scores compared
with the SAH + vehicle group (P<0.05; Fig. 4B). However, no significant
difference was revealed in neurological behavior between the SAH +
roscovitine (50 µg) and SAH + vehicle groups.
Roscovitine-induced increases in
neuronal survival after SAH
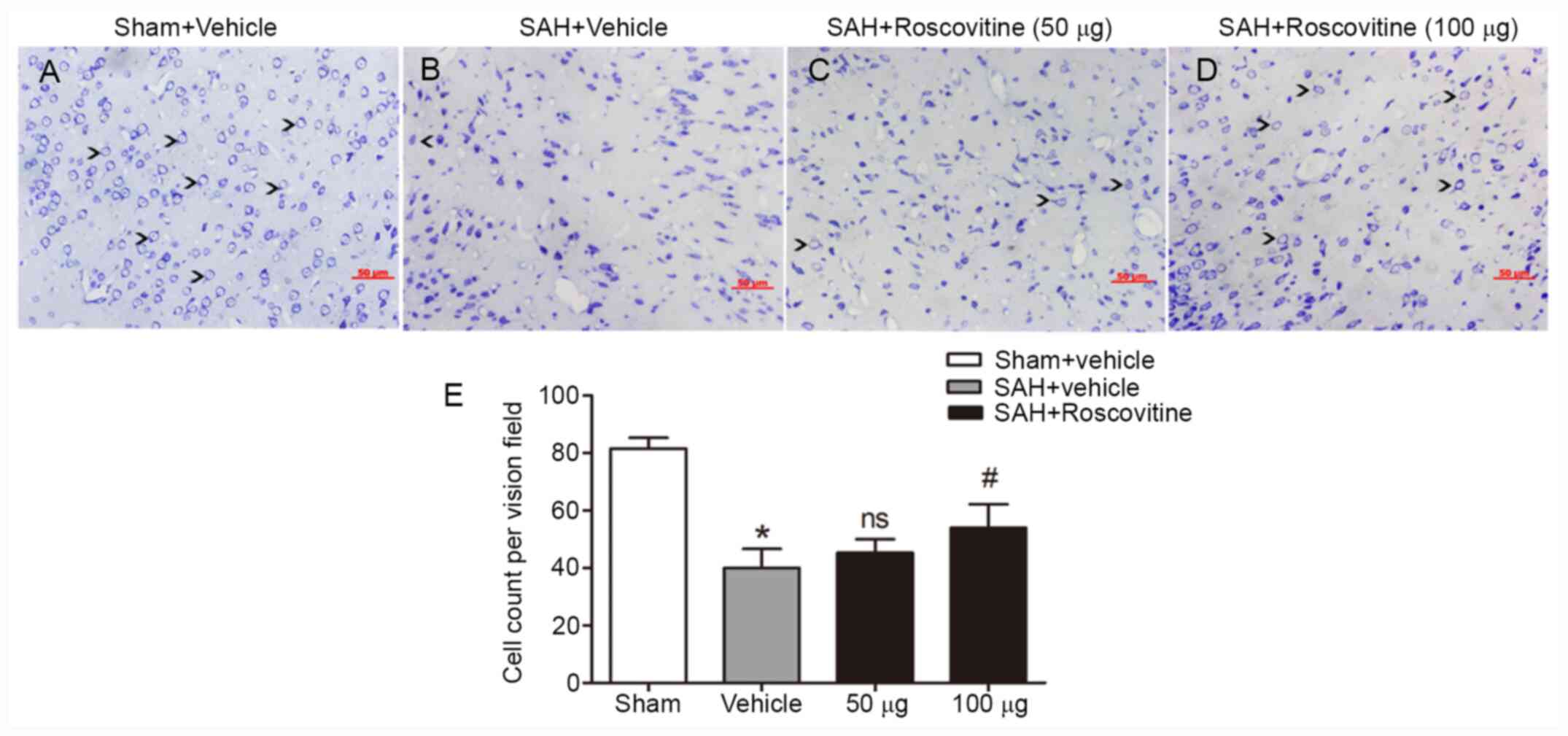
Nissl staining of the temporal cortex was performed
to explore the neuroprotective effect of roscovitine. The sham +
vehicle group exhibited neuronal cells with clear outline and
compact structures and cytoplasm (Fig.
5A). After SAH, the majority of neurons were damaged and
exhibited extensive degenerative changes. The neurons were sparsely
arranged, appeared to have lost their integrity, and exhibited
shrunken cytoplasm and swollen cell bodies (Fig. 5B-D). However, roscovitine (100 µg)
treatment significantly reduced the percentage of damaged neurons
in the SAH + roscovitine (100 µg) group compared with that in the
SAH + vehicle group (P<0.05; Fig.
5E).
SAH inhibits cortical neuronal
apoptosis after SAH
The dose of 100 µg of roscovitine was selected for
this experiment because it was determined to be most effective dose
based on the results of the above-described experiments. Few
TUNEL-positive neurons were found in the temporal cortex of the
sham + vehicle group (Fig. 6A-C).
The SAH + vehicle group demonstrated a significantly increased
percentage of apoptotic neurons compared with the sham + vehicle
group; however, roscovitine treatment (100 µg) significantly
reduced the percentage of TUNEL-positive cells in the SAH +
roscovitine (100 µg) group compared with the SAH + vehicle group
(Fig. 6D). These results indicated
that roscovitine treatment alleviated neuronal apoptosis after
SAH.
Effects of roscovitine on the
expression of cytochrome c and caspase-3
Compared with the sham + vehicle group, the SAH +
vehicle group displayed significantly increased expression levels
of cytochrome c and caspase-3 (P<0.05; Fig. 7A and B). However, cytochrome release and
caspase-3 cleavage were both significantly decreased after
roscovitine (100 µg) treatment in the SAH + roscovitine (100 µg)
group compared with the SAH + vehicle group (Fig. 7A and B).
Discussion
The present study determined that SAH led to
upregulation of the expression levels of Cdk5, p25 and Cdk5-pTyr15
proteins. Immunofluorescence staining highlighted the expression of
Cdk5 after SAH and that Cdk5 underwent nuclear translocation in
neurons. In addition, intracisternal administration of roscovitine
effectively improved neurological function, reduced the number of
apoptotic cells and alleviated brain edema. Furthermore, SAH
induced the upregulation of the expression levels of cytochrome c
and caspase-3, whereas roscovitine treatment significantly reduced
the expression of these proteins. Thus, the present study provided
strong evidence that Cdk5 was an important participant in the
development of EBI following SAH and may represent a focus for
potential SAH treatment in the future.
Previous studies confirmed that Cdk5 is activated in
several diseases of the nervous system, such as Alzheimer's disease
(30), cerebral ischemia (31) and Parkinson's disease (32). One regulatory mechanism of Cdk5
kinase activity is the phosphorylation of conserved residues
(33). Phosphorylation of Tyr15
residue of Cdk5 has a stimulatory effect and increases Cdk5 kinase
activity (14,34). In the present study, Cdk5 and
Cdk5-pTyr15 were significantly upregulated in the rat cortex
following SAH, indicating that Cdk5 kinase activity was increased.
In addition, as aberrant activation of Cdk5 requires the help of
the activator protein p25(35), the
protein levels of p25 were investigated and it was revealed that
p25 expression was elevated after SAH. p25 has a significantly
longer half-life compared with other proteins and can have
deleterious effects in neurons (30,36).
Increases in p25 protein expression can prolong Cdk5 activation.
The current study provided evidence of Cdk5 activation in the rat
brain after SAH.
A previous study suggested that Cdk5 is a regulator
of various neuronal cell death cascades (37). However, some studies have provided
contradictory evidence indicating that Cdk5 provides
neuroprotection in some conditions (38,39).
Several studies have revealed that the influence of Cdk5 on cell
death depended on its subcellular localization as opposed to its
activity. These studies suggested that Cdk5 plays a pro-survival
role in the cytoplasm but participates in death signaling within
the nucleus (40-42).
The present study suggested that Cdk5 was mostly expressed in the
cytoplasm in the sham group but translocated to the nucleus after
SAH, indicating that Cdk5 participates in neuronal cell death after
SAH.
Brain edema has been described as an accurate
predictor of EBI and an independent risk factor for unsatisfactory
outcomes in patients with SAH (43). Edema increases the mass and ICP
following SAH, which may lead to direct brain tissue damage and
ultimately, herniation. Mounting evidence and data has demonstrated
that apoptosis is strongly associated with the development of brain
edema after SAH (44,45). Endothelial cell apoptosis causes BBB
breakdown, thereby inducing brain edema. Moreover, neuronal
apoptosis contributes to cytotoxic brain edema, resulting in
subsequent neurological deficits (46,47).
To further elucidate the effects of Cdk5, the present study treated
SAH rats with roscovitine, a selective inhibitor of Cdk5. As
expected, roscovitine significantly alleviated brain edema after
SAH. Furthermore, roscovitine also reduced the number of apoptotic
cells and accelerated the recovery of neurological function
following SAH. Therefore, it is concluded that the inhibition of
Cdk5 alleviated brain edema and decreased neurobehavioral deficits
by mitigating neuronal apoptosis following SAH.
Accumulating evidence indicates that apoptosis is an
important intracellular pathway following SAH (48,49).
Although the precise mechanisms underlying apoptosis have not been
fully elucidated, mitochondrial dysfunction has been demonstrated
to be involved in apoptosis (40).
During apoptosis, the permeability of the outer mitochondrial
membrane increases, leading to the leakage of cytochrome c into the
cytosol. Interaction between cytochrome c and Apaf-1 facilitates
the formation of an apoptosome and activates caspase-3, leading to
neuronal apoptosis (50). Cdk5 is
involved in mitochondrial dysfunction; it triggers mitochondrial
fission and is an upstream regulator of the mitochondrial pathway
(51). Blocking Cdk5 activity
prevents mitochondria from releasing cytochrome c and preserves
mitochondrial integrity in injured neurons (52). The current study measured cytochrome
c and caspase-3 protein levels to further explore the mechanism by
which roscovitine exerts its anti-apoptotic effects. The results of
the present study suggested that the expression of cytochrome c and
caspase-3 substantially increased after SAH, which confirmed the
results of previous studies. However, treatment with roscovitine
(100 µg) significantly decreased the expression levels of
cytochrome c and caspase-3, indicating that roscovitine attenuates
neuronal apoptosis after SAH by inhibiting the expression levels of
cytochrome c and caspase-3, thereby reducing brain edema and
restoring neurological function.
The present study has certain limitations.
Roscovitine is not a specific inhibitor of Cdk5 because it also
inhibits Cdc2 and Cdk2(53).
However, previous studies have revealed that Cdk5 is expressed
mainly in post-mitotic neurons, whereas Cdc2 and Cdk2 are expressed
only at the embryonic stage (54,55).
Furthermore, all Cdk activity other than Cdk5 activity is
suppressed in mature neurons (56,57).
Kinase activity cannot be directly measured, so the content of
phosporylated-cdk5 was indirectly measured based on previous
literature (58). Therefore, in
conclusion, it is reasonable to speculate that the neuroprotective
effects noted in this experiment occurred mainly through the
inhibition of Cdk5 by roscovitine.
In summary, the results of the present study
demonstrated that SAH significantly upregulated the expression
levels of Cdk5, p25 and Cdk5-pTyr15 and induced apparent EBI. Cdk5
inhibition by roscovitine protected neurons from apoptosis
following SAH. Moreover, caspase-3 and cytochrome c were closely
associated with the anti-apoptotic effects of roscovitine on the
brain cells following experimental SAH. These data and results
demonstrated that Cdk5 plays a role in EBI after SAH and provides
support for the hypothesis that Cdk5 inhibitors can be developed as
therapeutic agents for SAH.
Acknowledgements
The authors would like to thank Dr Hua Li, Dr
Chun-Xi Wang and Dr Xiao-Ming Zhou from the Affiliated Suqian
Hospital of Xuzhou Medical University (Suqian, China) for providing
suggestions. In addition, the authors would like to acknowledge the
assistance provided by the following three pathologists: Dr
Qing-Song Wang, Dr Hai-Feng Liu and Dr Peng Zhao, Department of
Pathology of the Affiliated Suqian Hospital of Xuzhou Medical
University (Suqian, China).
Funding
Funding: This work was supported by The National Natural Science
Foundation of China (grant no. 81070974), The Jiangsu Provincial
Key Subject (grant no. X4200722) and The Jinling Hospital of
Nanjing, China (grant no. 2010Q017).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YD, LZ and JZ conceived and designed the project. LZ
and JZ confirm the authenticity of all the raw data and actually
performed the experiments. WZ, HL, XG and JLi contributed to
data/evidence collection and analysis. YD, LZ, JLiu, XN and JZ
analyzed and interpreted the data. JZ wrote the manuscript. All
authors have read and approved the final manuscript.
Ethics approval and consent to
participate
All study protocols regarding surgical procedures
and animal usage were approved by The Ethics Committee of the
Affiliated Suqian Hospital of Xuzhou Medical University (Suqian,
China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Connolly ES Jr, Rabinstein AA, Carhuapoma
JR, Derdeyn CP, Dion J, Higashida RT, Hoh BL, Kirkness CJ, Naidech
AM, Ogilvy CS, et al: Guidelines for the management of aneurysmal
subarachnoid hemorrhage: A guideline for healthcare professionals
from the American heart association/american stroke association.
Stroke. 43:1711–1737. 2012.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Hop JW, Rinkel GJ, Algra A and van Gijn J:
Case-fatality rates and functional outcome after subarachnoid
hemorrhage: A systematic review. Stroke. 28:660–664.
1997.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Mackey J, Khoury JC, Alwell K, Moomaw CJ,
Kissela BM, Flaherty ML, Adeoye O, Woo D, Ferioli S, De Los Rios La
Rosa F, et al: Stable incidence but declining case-fatality rates
of subarachnoid hemorrhage in a population. Neurology.
87:2192–2197. 2016.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Macdonald RL, Kassell NF, Mayer S,
Ruefenacht D, Schmiedek P, Weidauer S, Frey A, Roux S and Pasqualin
A: CONSCIOUS-1 Investigators. Clazosentan to overcome neurological
ischemia and infarction occurring after subarachnoid hemorrhage
(CONSCIOUS-1): Randomized, double-blind, placebo-controlled phase 2
dose-finding trial. Stroke. 39:3015–3021. 2008.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Sehba FA, Hou J, Pluta RM and Zhang JH:
The importance of early brain injury after subarachnoid hemorrhage.
Prog Neurobiol. 97:14–37. 2012.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Chaudhry SR, Kahlert UD, Kinfe TM, Endl E,
Dolf A, Niemelä M, Hänggi D and Muhammad S: Differential
polarization and activation dynamics of systemic T helper cell
subsets after aneurysmal subarachnoid hemorrhage (SAH) and during
post-SAH complications. Sci Rep. 11(14226)2021.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Liu L, Kawakita F, Fujimoto M, Nakano F,
Imanaka-Yoshida K, Yoshida T and Suzuki H: Role of periostin in
early brain injury after subarachnoid hemorrhage in mice. Stroke.
48:1108–1111. 2017.PubMed/NCBI View Article : Google Scholar : Suzuki H, Ayer R,
Sugawara T, Chen W, Sozen T, Hasegawa Y, Kanamaru K and Zhang JH:
Protective effects of recombinant osteopontin on early brain injury
after subarachnoid hemorrhage in rats. Crit Care Med 38: 612-618,
2010.
|
|
8
|
Uekawa K, Hasegawa Y, Ma M, Nakagawa T,
Katayama T, Sueta D, Toyama K, Kataoka K, Koibuchi N, Kawano T, et
al: Rosuvastatin ameliorates early brain injury after subarachnoid
hemorrhage via suppression of superoxide formation and nuclear
factor-kappa B activation in rats. J Stroke Cerebrovasc Dis.
23:1429–1439. 2014.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Feng D, Wang W, Dong Y, Wu L, Huang J, Ma
Y, Zhang Z, Wu S, Gao G and Qin H: Ceftriaxone alleviates early
brain injury after subarachnoid hemorrhage by increasing excitatory
amino acid transporter 2 expression via the PI3K/Akt/NF-κB
signaling pathway. Neuroscience. 268:21–32. 2014.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Wang Z, Guo S, Wang J, Shen Y, Zhang J and
Wu Q: Nrf2/HO-1 mediates the neuroprotective effect of mangiferin
on early brain injury after subarachnoid hemorrhage by attenuating
mitochondria-related apoptosis and neuroinflammation. Sci Rep.
7(11883)2017.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Mo J, Enkhjargal B, Travis ZD, Zhou K, Wu
P, Zhang G, Zhu Q, Zhang T, Peng J, Xu W, et al: AVE 0991
attenuates oxidative stress and neuronal apoptosis via
Mas/PKA/CREB/UCP-2 pathway after subarachnoid hemorrhage in rats.
Redox Biol. 20:75–86. 2019.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Pang J, Chen Y, Kuai L, Yang P, Peng J, Wu
Y, Chen Y, Vitek MP, Chen L, Sun X and Jiang Y: Inhibition of
blood-brain barrier disruption by an apolipoprotein E-mimetic
peptide ameliorates early brain injury in experimental subarachnoid
hemorrhage. Transl Stroke Res. 8:257–272. 2017.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Mangold N, Pippin J, Unnersjoe-Jess D,
Koehler S, Shankland S, Brähler S, Schermer B, Benzing T,
Brinkkoetter PT and Hagmann H: The atypical cyclin-dependent kinase
5 (Cdk5) guards podocytes from apoptosis in glomerular disease
while being dispensable for podocyte development. Cells.
10(2464)2021.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Roufayel R and Murshid N: CDK5: Key
regulator of apoptosis and cell survival. Biomedicines.
7(88)2019.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Shah K and Lahiri DK: A tale of the good
and bad: Remodeling of the microtubule network in the brain by
Cdk5. Mol Neurobiol. 54:2255–2268. 2017.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Sharma S and Sicinski P: . A kinase of
many talents: Non-neuronal functions of CDK5 in development and
disease. Open Biol. 10(190287)2020.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Mushtaq G, Greig NH, Anwar F, Al-Abbasi
FA, Zamzami MA, Al-Talhi HA and Kamal MA: Neuroprotective
mechanisms mediated by CDK5 inhibition. Curr Pharm Des. 22:527–534.
2016.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Menn B, Bach S, Blevins TL, Campbell M,
Meijer L and Timsit S: Delayed treatment with systemic
(S)-roscovitine provides neuroprotection and inhibits in vivo CDK5
activity increase in animal stroke models. PLoS One.
5(e12117)2010.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Tuo QZ, Liuyang ZY, Lei P, Yan X, Shentu
YP, Liang JW, Zhou H, Pei L, Xiong Y, Hou TY, et al: Zinc induces
CDK5 activation and neuronal death through CDK5-Tyr15
phosphorylation in ischemic stroke. Cell Death Dis.
9(870)2018.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Zhou YF, Wang J, Deng MF, Chi B, Wei N,
Chen JG, Liu D, Yin X, Lu Y and Zhu LQ: The peptide-directed
lysosomal degradation of CDK5 exerts therapeutic effects against
stroke. Aging Dis. 10:1140–1145. 2019.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Kumar S, Rajput MK and Tickoo SB: Laws,
regulations, policies and guidelines governing the care and use of
laboratory animals. Essentials of laboratory animal science:
Principles and practices. Springer, Singapore, pp23-38, 2021.
|
|
22
|
Sun Q, Dai Y, Zhang X, Hu YC, Zhang D, Li
W, Zhang XS, Zhu JH, Zhou ML and Hang CH: Expression and cell
distribution of myeloid differentiation primary response protein 88
in the cerebral cortex following experimental subarachnoid
hemorrhage in rats: A pilot study. Brain Res. 1520:134–144.
2013.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Wang KC, Tang SC, Lee JE, Tsai JC, Lai DM,
Lin WC, Lin CP, Tu YK and Hsieh ST: Impaired microcirculation after
subarachnoid hemorrhage in an in vivo animal model. Sci Rep.
8(13315)2018.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Li R, Liu W, Yin J, Chen Y, Guo S, Fan H,
Li X, Zhang X, He X and Duan C: TSG-6 attenuates
inflammation-induced brain injury via modulation of microglial
polarization in SAH rats through the SOCS3/STAT3 pathway. J
Neuroinflammation. 15(231)2018.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Lee JY, Sagher O, Keep R, Hua Y and Xi G:
Comparison of experimental rat models of early brain injury after
subarachnoid hemorrhage. Neurosurgery. 65:331–343. 2009.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Hilton GD, Stoica BA, Byrnes KR and Faden
AI: Roscovitine reduces neuronal loss, glial activation, and
neurologic deficits after brain trauma. J Cereb Blood Flow Metab.
28:1845–1859. 2008.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Nicolás P, Lassalle VL and Ferreira ML:
Quantification of immobilized Candida antarctica lipase B (CALB)
using ICP-AES combined with Bradford method. Enzyme Microb Technol.
97:97–103. 2017.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Zhuang Z, Zhou ML, You WC, Zhu L, Ma CY,
Sun XJ and Shi JX: Hydrogen-rich saline alleviates early brain
injury via reducing oxidative stress and brain edema following
experimental subarachnoid hemorrhage in rabbits. BMC Neurosci.
13(47)2012.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Garcia JH, Wagner S, Liu KF and Hu XJ:
Neurological deficit and extent of neuronal necrosis attributable
to middle cerebral artery occlusion in rats. Statistical
validation. Stroke. 26:627–635. 1995.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Liu X, Feng Z, Du L, Huang Y, Ge J, Deng Y
and Mei Z: The potential role of MicroRNA-124 in cerebral ischemia
injury. Int J Mol Sci. 21(120)2019.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Cheng X, Xu S, Zhang C, Qin K, Yan J and
Shao X: The BRCC3 regulated by Cdk5 promotes the activation of
neuronal NLRP3 inflammasome in Parkinson's disease models. Biochem
Biophys Res Commun. 522:647–654. 2020.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Ji YB, Zhuang PP, Ji Z, Wu YM, Gu Y, Gao
XY, Pan SY and Hu YF: TFP5 peptide, derived from CDK5-activating
cofactor p35, provides neuroprotection in early-stage of adult
ischemic stroke. Sci Rep. 7(40013)2017.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Shupp A, Casimiro MC and Pestell RG:
Biological functions of CDK5 and potential CDK5 targeted clinical
treatments. Oncotarget. 8:17373–17382. 2017.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Zhang B, Tan VB, Lim KM and Tay TE: The
activation and inhibition of cyclin-dependent kinase-5 by
phosphorylation. Biochemistry. 46:10841–10851. 2007.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Ko YU, Kim C, Lee J, Kim D, Kim Y, Yun N
and Oh YJ: Site-specific phosphorylation of Fbxw7 by Cdk5/p25 and
its resulting decreased stability are linked to glutamate-induced
excitotoxicity. Cell Death Dis. 10(579)2019.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Lee MS, Kwon YT, Li M, Peng J, Friedlander
RM and Tsai LH: Neurotoxicity induces cleavage of p35 to p25 by
calpain. Nature. 405:360–364. 2000.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Zhang H, Chang L, Zhang H, Nie J, Zhang Z,
Yang X, Vuong AM, Wang Z, Chen A and Niu Q: Calpain-2/p35-p25/Cdk5
pathway is involved in the neuronal apoptosis induced by
polybrominated diphenyl ether-153. Toxicol Lett. 277:41–53.
2017.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Tucker D, Lu Y and Zhang Q: From
mitochondrial function to neuroprotection-an emerging role for
methylene blue. Mol Neurobiol. 55:5137–5153. 2018.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Sénécal V, Barat C and Tremblay MJ: The
delicate balance between neurotoxicity and neuroprotection in the
context of HIV-1 infection. Glia. 69:255–280. 2021.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Tiernan CT, Ginsberg SD, He B, Ward SM,
Guillozet-Bongaarts AL, Kanaan NM, Mufson EJ and Counts SE:
Pretangle pathology within cholinergic nucleus basalis neurons
coincides with neurotrophic and neurotransmitter receptor gene
dysregulation during the progression of Alzheimer's disease.
Neurobiol Dis. 117:125–136. 2018.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Sun YQ, Xie JW, Xie HT, Chen PC, Zhang XL,
Zheng CH, Li P, Wang JB, Lin JX, Cao LL, et al: Expression of CRM1
and CDK5 shows high prognostic accuracy for gastric cancer. World J
Gastroenterol. 23:2012–2022. 2017.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Ruegsegger GN, Toedebusch RG, Childs TE,
Grigsby KB and Booth FW: Loss of Cdk5 function in the nucleus
accumbens decreases wheel running and may mediate age-related
declines in voluntary physical activity. J Physiol. 595:363–384.
2017.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Tanabe J, Nakahara I, Matsumoto S, Suyama
Y, Morioka J, Oda J, Hasebe A, Suzuki T, Watanabe S, Suyama K, et
al: Cortical blood flow insufficiency scores with computed
tomography perfusion can predict outcomes in aneurysmal
subarachnoid hemorrhage patients: A cohort study. Neurocrit Care.
34:946–955. 2021.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Qian C, Jin J, Chen J, Li J, Yu X, Mo H
and Chen G: SIRT1 activation by resveratrol reduces brain edema and
neuronal apoptosis in an experimental rat subarachnoid hemorrhage
model. Mol Med Rep. 16:9627–9635. 2017.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Shamsi Meymandi M, Soltani Z, Sepehri G,
Amiresmaili S, Farahani F and Moeini Aghtaei M: Effects of
pregabalin on brain edema, neurologic and histologic outcomes in
experimental traumatic brain injury. Brain Res Bull. 140:169–175.
2018.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Liu XC, Wu CZ, Hu XF, Wang TL, Jin XP, Ke
SF, Wang E and Wu G: Gastrodin attenuates neuronal apoptosis and
neurological deficits after experimental intracerebral hemorrhage.
J Stroke Cerebrovasc Dis. 29(104483)2020.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Sadeghian N, Shadman J, Moradi A, Ghasem
Golmohammadi M and Panahpour H: Calcitriol protects the blood-brain
barrier integrity against ischemic stroke and reduces vasogenic
brain edema via antioxidant and antiapoptotic actions in rats.
Brain Res Bull. 150:281–289. 2019.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Sun J and Nan G: The extracellular
signal-regulated kinase 1/2 pathway in neurological diseases: A
potential therapeutic target (review). Int J Mol Med. 39:1338–1346.
2017.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Huang YH, Chung CL, Tsai HP, Tzou RD, Wu
SC, Chai CY, Lee TC and Kwan AL: Impact of hyperglycemia on
neuronal apoptosis after subarachnoid hemorrhage in rodent brain:
An experimental research. Int J Surg. 83:246–252. 2020.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Yadav N, Gogada R, O'Malley J, Gundampati
RK, Jayanthi S, Hashmi S, Lella R, Zhang D, Wang J, Kumar R, et al:
Molecular insights on cytochrome c and nucleotide regulation of
apoptosome function and its implication in cancer. Biochim Biophys
Acta Mol Cell Res. 1867(118573)2020.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Guo MY, Shang L, Hu YY, Jiang LP, Wan YY,
Zhou QQ, Zhang K, Liao HF, Yi JL and Han XJ: The role of
Cdk5-mediated Drp1 phosphorylation in Aβ1-42 induced
mitochondrial fission and neuronal apoptosis. J Cell Biochem.
119:4815–4825. 2018.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Seager R, Lee L, Henley JM and Wilkinson
KA: Mechanisms and roles of mitochondrial localisation and dynamics
in neuronal function. Neuronal Signal. 4(NS20200008)2020.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Kim JE, Park H, Choi SH, Kong MJ and Kang
TC: Roscovitine attenuates microglia activation and monocyte
infiltration via p38 MAPK inhibition in the rat frontoparietal
cortex following status epilepticus. Cells. 8(746)2019.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Kartha CC: Cell cycle regulation in
cardiomyocytes. In: Cardiomyocytes in health and disease. Springer,
Cham, pp25-39, 2021.
|
|
55
|
Chao AC, Chen CH, Wu MH, Hou BY and Yang
DI: Roles of Id1/HIF-1 and CDK5/HIF-1 in cell cycle reentry induced
by amyloid-beta peptide in post-mitotic cortical neuron. Biochim
Biophys Acta Mol Cell Res. 1867(118628)2020.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Cortés N, Guzmán-Martínez L, Andrade V,
González A and Maccioni RB: CDK5: A unique CDK and its multiple
roles in the nervous system. J Alzheimers Dis. 68:843–855.
2019.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Chen SD, Yang JL, Lin YC, Chao AC and Yang
DI: Emerging roles of inhibitor of differentiation-1 in Alzheimer's
disease: Cell cycle reentry and beyond. Cells.
9(1746)2020.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Lee J, Ko YU, Chung Y, Yun N, Kim M, Kim K
and Oh YJ: The acetylation of cyclin-dependent kinase 5 at lysine
33 regulates kinase activity and neurite length in hippocampal
neurons. Sci Rep. 8(13676)2018.PubMed/NCBI View Article : Google Scholar
|