Introduction
Severe aplastic anemia (SAA) is a rare and
potentially fatal disease characterized by pancytopenia and bone
marrow (BM) hypoplasia (1).
Patients frequently experience symptoms such as anemia, bleeding
and infection, leading to a significant decrease in quality of life
and eventually death (2). The
abnormal activation and hyperfunction of hyper-activated effector T
lymphocytes are the major immune mechanisms in the pathogenesis of
primary acquired SAA (3,4). However, the etiology and pathogenesis
of SAA have remained to be fully elucidated. Previous research by
our group has indicated that SAA is caused by an unknown antigen
substance activating dendritic cells, which increase the number and
function of myeloid dendritic cells (mDCs), resulting in the
hyperfunction of cytotoxic T lymphocyte (CTL) and T helper type 1
(Th1) cells. This causes the release of numerous negative
hematopoietic regulatory factors. The regulatory T cells and
natural killer (NK) cells that induce immune tolerance are
consequently severely insufficient and damage hematopoietic target
cells, leading to hematopoietic failure (5,6). In
the previous study, mDCs proteomes were investigated using mass
spectrometry and the CD34+ cells using isobaric tags for
relative and absolute quantification (iTRAQ) labeling and a
combination of multidimensional liquid chromatography and tandem
mass spectrometry (7). In
comparison with healthy controls, the expression of leukocyte
immunoglobulin-like receptors eukocyte immunoglobulin-like receptor
A3 (LILRA3) in mDCs and LILRA5 in CD34+ cells were
significantly increased.
LILRs are members of the immunoglobulin superfamily
that are mainly expressed on antigen-presenting cells (APCs). The
genes are located on chromosome 19q13.4 (8,9). They
are divided into immunosuppressive receptor LILRs (LILRB1-5) and
immunoreactive receptor LILRs (LILRA1-6) according to the
cytoplasmic structures and transmembrane regions (10). After binding to major
histocompatibility complex I (MHC-I) ligand, it changes the
autoimmune-mediated tissue damage threshold and regulates the
immune responses through the inhibition or activation of cytolysis.
MHC-I molecules are widely expressed on hematopoietic cells of all
lineages. When the intracellular protein fragment-short antigen
peptide is presented by CTL, the LILRA-MHC-I interaction
effectively stimulates T-cell proliferation and secretes
inflammatory factors (11).
Enhancing CTL lysis clears target cells expressing autoantigens,
making the LILR a novel target for regulating CTL-mediated
autoimmune diseases (12,13). LILRs are associated with autoimmune
diseases, malignancies and infections (14,15).
However, little is currently known about the potential role of
LILRA genes in patients with SAA. The present study aimed to
analyze the expression of LILRAs in patients with SAA and
investigate its role in the pathogenesis of SAA by determining its
impact on patient-derived mDCs and targeted BM CD34+
cells.
Materials and methods
Patients
A total of 48 patients with SAA that were admitted
to the Hematology Department of Tianjin Medical University General
Hospital (Tianjin, China) between June 2015 and April 2017 were
enrolled. Of these, 26 were newly diagnosed, untreated patients
with SAA (12 males, 14 females) with a median age of 28 years
(range, 7-69 years). The other 22 patients were remission-treated
patients with SAA (14 males, 8 females) with a median age of 31
years (range, 11-65 years). Patients in remission were those who
improved after immunosuppressive therapy (IST, including
antithymocyte globulin, cyclosporine and glucocorticoid). All the
patients in remission had achieved bone marrow hematopoietic
recovery and had been separated from the infusion of blood
products, while some had normal peripheral blood cell counts but
still required drug therapy. All subjects met the diagnostic
criteria (16,17). Patients were excluded if they had
complications such as iron overload, malignancy or other autoimmune
diseases or if they were pregnant. The patients' clinical
characteristics are provided in Table
I.
 | Table IClinical characteristics of the
patients. |
Table I
Clinical characteristics of the
patients.
| Item | Untreated SAA
(n=26) | R-SAA (n=22) |
|---|
| Age (years) | 28 (7-69) | 31 (11-65) |
| ANC
(x109/l) | 0.58±0.38 | 3.01±1.76 |
| Hb (g/l) | 58.14±23.32 | 127.19±38.64 |
| PLT
(x109/l) | 18.31±11.62 | 118.84±70.31 |
| Ret% | 0.41±0.29 | 2.89±1.97 |
| Therapy | Not previously
treated except for transfusions | IST |
| Duration
(months) | 2 (1-3) | 30 (7-109) |
All patients received immunosuppressive therapy that
included rabbit anti-thymocyte globulin, cyclosporin and
hematopoietic stimulating factor treatment consisting of
erythropoietin, granulocyte colony stimulating factor,
thrombopoietin and interleukin IL-11. Treatment efficacy was
evaluated based on the Camitta standard (18).
The healthy controls (13 males and 15 females)
included had a median age of 28 years (range, 24-55 years). The
present study was approved by the Ethics Committee of Tianjin
Medical University (Tianjin, China). Written informed consent was
obtained from all study subjects.
Culture, identification and sorting of
mDCs
BM mononuclear cells (BMMNCs) were extracted from
patients with SAA and healthy controls using lymphocyte separation
fluid (Solarbio Science & Technology) using density gradient
centrifugation. BMMNCs of each subject were plated separately at a
density of 2x106 cells/ml in RPMI-1640 (Gibco; Thermo
Fisher Scientific, Inc.) complete medium containing 10% fetal
bovine serum (Gibco; Thermo Fisher Scientific, Inc.) and 1%
mycillin and incubated for 2 h at 37˚C in an atmosphere containing
5% CO2. Non-adherent cells were removed. The remaining
cells were cultured in RPMI-1640 complete medium containing 10%
fetal bovine serum, 1% mycillin, 100 ng/ml Recombinant Human
Granulocyte-Macrophage Colony Stimulating Factor (cat. no.
10015-HNAH; Sino Biological) and 40 ng/ml rhIL-4. The culture
conditions were 37˚C and 5% CO2. Media and cytokines
were changed every two days. On day 6, rhTNF-α (1,000 µg/ml) was
added to mature the mDCs for 24 h. Suspended mature mDCs in the
culture supernatant were then collected on day 7. The collected
cells were stained with PerCP-conjugated human leukocyte antigen
(HLA)-DR (cat. no. 347364) and APC-conjugated CD11c monoclonal
antibody (cat. no. 340544; all, BD Biosciences).
HLA-DR+CD11c+ cells were sorted and collected
using a FACS Aria flow cytometer (BD Biosciences).
Reverse transcription-quantitative
(RT-q)PCR
The mRNA expression of LILRA was analyzed using
RT-qPCR. BMMNCs of patients with SAA and controls were lysed using
TRIzol reagent. RNA was reverse transcribed using the complementary
(c)DNA Synthesis Kit (Tiangen). qPCR was performed on the BIORAD
iQ5 system (Bio-Rad Laboratories, Inc.). GAPDH was used as a
housekeeping gene for standardizing targeted mRNA expression. A
total of 1 µl of each cDNA working solution was used with a final
volume of 25 µl, which contained 12.5 µl SYBR green solution and
0.75 µl of upstream and downstream primers (concentration, 10 µM).
PCR reaction conditions were as follows: LILRA1-6: 95˚C for 30 sec,
the indicated annealing temperature for 45 sec, 72˚C for 30 sec, 45
cycles. The indicated annealing temperature and primer sequences
are listed in Table II. The
relative expression level of the gene of interest was calculated
using the 2-ΔΔCq method (19).
| Table IISequences of PCR primers and The
indicated annealing temperature. |
Table II
Sequences of PCR primers and The
indicated annealing temperature.
| Gene | Sequence (5' to
3') | Annealing
temperature (˚C) |
|---|
| LILRA1 | Forward:
5'-CCTCGGGATTCTGCTATTTG-3' | 62.0 |
| | Reverse:
5'-AAGGCTCCACCACTCTGAAG-3 | |
| LILRA2 | Forward:
5'-TGGGGACCTACAGATGCTACA-3' | 55.5 |
| | Reverse:
5'-CTTGTTTTGTGATGGGCTGA-3' | |
| LILRA3 | Forward:
5'-CAGCCCACCACAAAACAAG-3' | 61.5 |
| | Reverse:
5'-CTTCAAATGTCCACCCAGGA-3' | |
| LILRA4 | Forward:
5'-AGGAGGCAAACAGCAGAAAG-3' | 62.0 |
| | Reverse:
5'-CAGCAGACACTTCCCCAACT-3' | |
| LILRA5 | Forward:
5'-TCTGACTGAGGAAGGAGACCA-3' | 57.0 |
| | Reverse:
5'-CCATAGCATCTGAGCATCCA-3' | |
| LILRA6 | Forward:
5'-ACCTGCTGTCTTTCCCCAGT-3' | 60.0 |
| | Reverse:
5'-TGTGTAATCCTTGGCGTGTG-3' | |
| GAPDH | Forward:
5'-TTCCACCCATGGCAAATTCC-3' | |
| | Reverse:
5'-AGGCCATGCCAGTGAGCTTC-3' | |
Flow cytometric (FCM) analysis
For phenotype analysis, fresh heparinized BM samples
were stained with anti-human LILRA3-FITC (cat. no. IC2574G),
LILRA5-PE monoclonal antibody (mAb; cat. no. FAB6754P; both from
R&D Systems), CD34-PerCP (cat. no. 340430), CD3-APC/PerCP (cat.
no. 340440 or 347344), CD8-APC/PerCP (cat. no. 340584 or 347314),
CD19-APC (cat. no. 340437), CD14-PE/FITC mAb (cat. no. 347497 or
347493), CD11c-APC (cat. no. 340544) and HLA-DR-PerCP mAb (cat. no.
347364; all from BD Biosciences). The staining was performed
according to the manufacturer's protocol. The ratios of
intracytoplasmic
LILRA3+CD11c+HLA-DR+/CD11c+HLA-DR+,
LILRA3+CD34+/CD34+,
LILRA3+CD3+CD8+/CD3+CD8+,
LILRA3+CD19+/CD19+,
LILRA3+CD14+/CD14+,
LILRA5+CD11c+HLA-DR+/CD11c+HLA-DR+,
LILRA5+CD34+/CD34+,
LILRA5+CD3+CD8+/CD3+CD8+,
LILRA5+CD19+/CD19+ and
LILRA5+CD14+/CD14+ cells were
analyzed on a FACS Calibur flow cytometer (BD Biosciences) and
CellQuest software version 3.1 software (BD Biosciences).
Western blot analysis
The mDCs of the subjects from the SAA, R-SAA and
healthy control groups were collected and lysed in RIPA buffer
supplemented with PMSF. The protein concentration was measured
using a BCA kit. The loading volume per sample was 40 µg protein.
The proteins were separated on 10% SDS-PAGE (Beijing Solarbio
Science & Technology Co., Ltd.) and transferred to a
nitrocellulose membrane. The membrane was blocked with 10% skimmed
milk (BD Biosciences; cat. no. 232100 for 1 h at room temperature.
The membrane was stained with anti-LILRA3 (dilution, 1:1,000, cat.
no. ab111562; Abcam) and anti-β-actin antibodies (dilution,
1:1,000; cat. no. 4970; Cell Signaling Technology, Inc.) for 12 h
at 4˚C. After washes with tris-buffered saline containing Tween-20,
the membrane was incubated with a secondary antibody (cat. no.
ZB-2305; dilution, 1:10,000; goat anti-mouse, Zhongshanjinqiao) for
1 h at room temperature. The bands were visualized with the Super
ECL Plus Detection Reagent (Applygen Technologies Inc.). Protein
levels were normalized to β-actin.
ELISA
Serum levels of soluble LILRA3 were measured using
an ELISA reagent kit (cat. no. SEB387Hu; Cloud Clone). According to
the manufacturer's protocols, the samples were measured and read
with a BioTek ELx800 microplate reader (BioTek Corp.) at a
wavelength of 450 nm.
Cytokine detection
A cytokine detection kit (Human Th1/Th2 subsets
detection kit) was used for this analysis (cat. no. P010001;
Saijishengwu). The venous blood samples were collected in EDTA
anticoagulation tubes and centrifuged at 1,000 x g for 20 min for
later use. Standards were configured with the following
concentrations: 10, 20, 40, 80, 156, 312, 625, 1,250, 2,500 and
5,000 pg/ml. The captured microsphere mixture was centrifuged at
200 x g for 5 min at room temperature. The supernatant was
aspirated, the same volume of microsphere buffer as the aspirated
supernatant was added, and following thorough mixing, the sample
was incubated for 30 min at room temperature. Subsequently, 25 µl
of the solution was added to each experimental tube and the sample
was vortexed. A total of 25 µl of standard product and 25 µl of the
sample to be tested was added in the same sample tube.
Subsequently, 25 µl of the fluorescence detection reagent was added
to each experimental tube and samples were thoroughly mixed and
incubated at room temperature for 25 min in the dark. Finally, 1 ml
PBS was added to each experimental tube, which was then centrifuged
at 200 x g for 5 min at room temperature. The supernatant was
aspirated and 100 µl PBS was added for fluorescence detection on
the flow cytometer (BD Biosciences; FACSCanto II). This was
performed according to the manufacturer's instructions.
Statistical analysis
Statistical analyses were performed using SPSS 22.0
software (IBM Corp.). Data analyses were performed with GraphPad
Prism 5.0 software (GraphPad Software, Inc.). The normality of the
distribution was proven using a Kolmogorov-Smirnov test. The mean ±
standard deviation was used to represent normally distributed data.
Comparisons between two independent samples were performed using
the t-test. The rank-sum test was used to analyze data with a
non-normal distribution and the median and interquartile range were
used to represent the data. Age and duration values are mid (min,
max). For correlation tests, Spearman's rank correlation was used.
P<0.05 was considered to indicate a statistically significant
difference.
Results
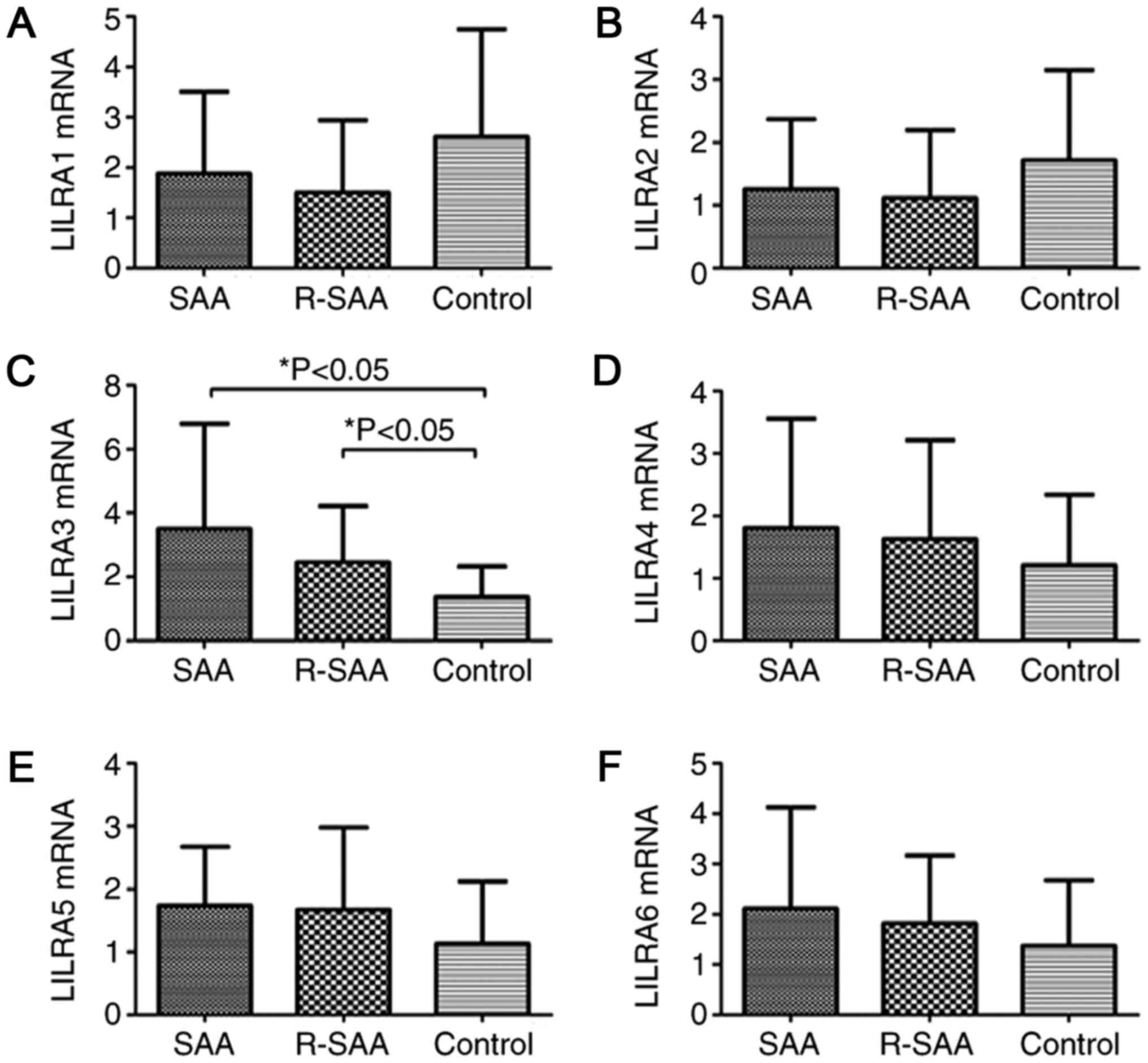
Increased LILRA3 mRNA expression in
patients with SAA
The expression of transcription factor LILRA1-6 in
patients with SAA and healthy controls was assessed using RT-qPCR.
The results suggested that the relative expression of LILRA3 mRNA
of untreated patients with SAA (3.513±3.291) and remission patients
with SAA (2.451±1.767) were significantly higher than that of the
controls (1.372±0.961; P<0.05). There was no significant
difference between the untreated and remission groups (P>0.05).
The relative expression of LILRA5 mRNA in untreated patients with
SAA (1.738±0.935) and remission patients with SAA (1.671±1.308) was
higher than that in the controls (1.128±0.993); however, the
differences were not statistically significant. The relative
expression levels of LILRA1 (1.880±1.627 vs. 1.502±1.438 vs.
2.609±2.144; P>0.05), LILRA2 (1.255±1.114 vs. 1.113±1.083 vs.
1.714±1.437; P>0.05), LILRA4 (1.808±1.751 vs. 1.625±1.588 vs.
1.213±1.124; P>0.05) and LILRA6 (2.111±2.018 vs. 1.813±1.357 vs.
1.371±1.302; P>0.05) were not significantly different between
patients with SAA and the R-SAA and controls (Fig. 1).
Increased frequency of LILRA3 on BM
mDCs in patients with SAA
The percentage of LILRA3 and LILRA5 on
CD34+ cells, CD3+ cells, CD8+
cells and CD11C+ HLA-DR+ cells from 48
patients with SAA and 28 healthy controls was analyzed by FCM
(Fig. 2A). The fraction of
intracytoplasmic LILRA3+ cells among mDCs of untreated
patients with SAA (73.11±22.54%) was significantly higher than that
of the controls (48.64±38.03%; P<0.05). There were no
significant differences in the percentages between the remission
patients with SAA (60.22±23.69%) and controls (P>0.05; Fig. 2B). The proportion of
LILRA5+ cells among mDCs, the relative expression of
LILRA5 mRNA and the relative intensity of LILRA5 in BM mDCs were
not significantly different between patients with SAA and healthy
controls (P>0.05; Fig. 2B). mDCs
were isolated from the BM of patients with SAA and healthy
controls, and the purity of sorted mDCs was >90% (Fig. 3A). The results further suggested
that the relative expression of LILRA3 mRNA in BM mDCs of untreated
patients with SAA (1.792±1.301) was higher than that of the
controls (0.773±0.721; P<0.05). In comparison with the controls,
the relative intensity of LILRA3 protein in mDCs of the untreated
SAA group was significantly increased (P<0.05; Fig. 3B).
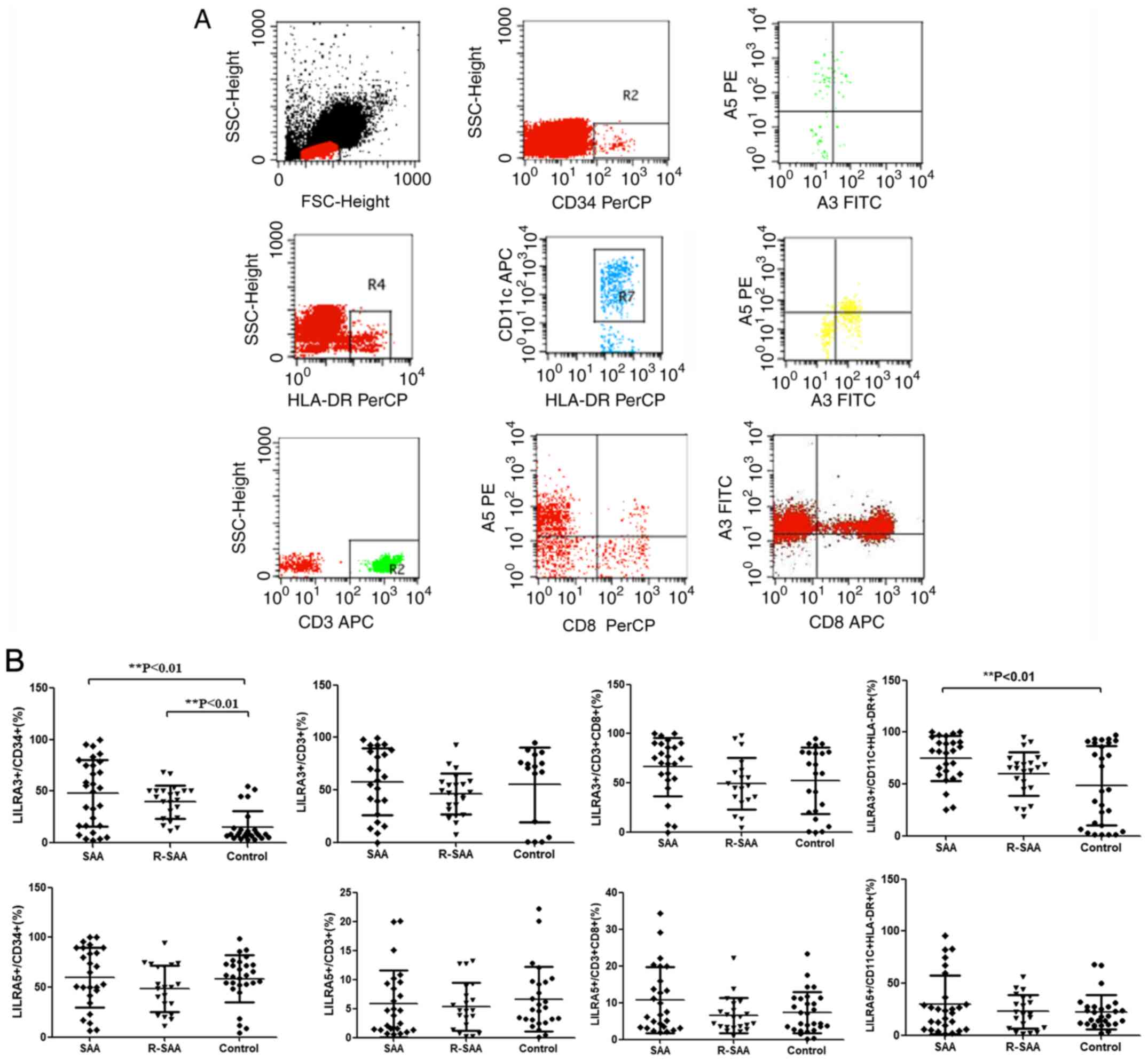 | Figure 2Expression of LILRA3 and LILRA5 on
CD34+ cells, CD3+ cells, CD8+
cells and CD11C+ HLA-DR+ cells. (A)
Expression assessed using flow cytometry. (B) LILRA3 and LILRA5
expression on CD34+, CD3+, CD8+
and CD11C+ HLA-DR+ cells in patients with SAA
and normal controls. LILRA, leukocyte immunoglobulin-like receptors
A; R-SAA, remission-treated severe aplastic anemia; PLT, platelets;
HB, hemoglobin; HLA, human leukocyte antigen; SSC, side
scatter. |
Increased percentage of LILRA3 on BM
CD34+ cells in patients with SAA
Untreated (47.05±32.07%) and remission patients with
SAA (39.86±19.46%) had increased percentages of intracytoplasmic
LILRA3+CD34+ cells compared with the controls
(14.26±14.00%; P<0.05). The percentages of BM LILRA5+
CD34+ cells were not different between patients with SAA
and healthy controls (P>0.05). Furthermore, the percentages of
LILRA3+ and LILRA5+ cells among the
CD14+, CD19+ and CD8+ lymphocytes
in patients with SAA were not different compared with those of the
healthy controls (P>0.05; data not shown).
Increased serum LILRA3 levels in
patients with SAA
Patients with SAA had increased serum levels of
LILRA3 in comparison with those of the controls. The serum LILRA3
levels of untreated patients with SAA (9.466±2.629 ng/ml) were
significantly higher than those of the remission patients with SAA
(4.591±2.958 ng/ml; P<0.05) and controls (3.682±2.695 ng/ml;
P<0.05; Fig. 3B).
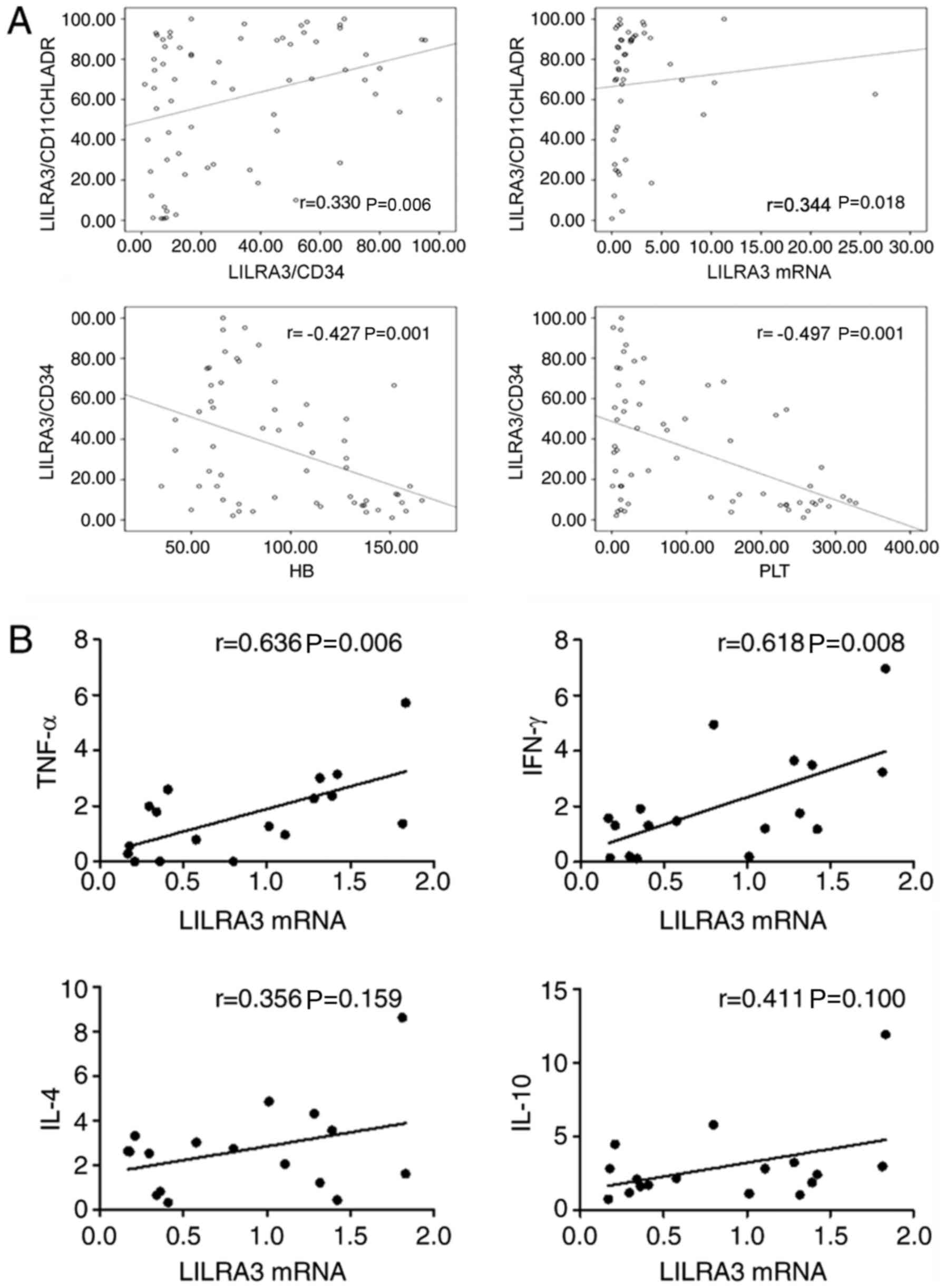
Frequency of LILRA3 is closely
associated with clinical characteristics and cytokine levels in
patients with SAA
To determine whether the frequency of LILRA3 may
identify a pathogenically distinct subset of patients with SAA or
whether it simply reflects disease activity, clinical indices and
evidence of disease activity in BM were compared with the
expression of LILRA3.
In the SAA group, there were positive correlations
between the proportion of
LILRA3+CD11C+HLA-DR+/CD11C+HLA-DR+
cells and the ratio of
LILRA3+CD34+/CD34+, the expression
of LILRA3 mRNA (r=0.330 and 0.344, respectively; P<0.05). There
were negative correlations between the ratio of LILRA3+
CD34+/CD34+ and the platelet count and
hemoglobin levels (r=-0.497 and -0.427, respectively; P<0.05;
Fig. 4A).
The levels of LILRA3 mRNA and serum cytokine TNF-α
(r=0.636, P<0.05) and IFN-γ (r=0.618, P<0.05) in untreated
patients with SAA were positively correlated. There was no
significant correlation between LILRA3 mRNA levels and serum
cytokine IL-4 (r=0.356, P>0.05) or IL-10 (r=0.411, P>0.05) in
untreated patients with SAA (Fig.
4B).
Discussion
The present study was the first to indicate an
alteration of BM LILRAs in patients with SAA. SAA is a type of BM
failure mediated by abnormal cellular immunity. To date, the
etiology and pathogenesis of SAA have remained to be fully
elucidated. Previous studies by our group have confirmed that
various immune cells and cytokines constitute an abnormal immune
status in patients with SAA (20,21),
including the involvement of Th1/Th2 subset imbalances,
hyperfunctional CTLs, insufficiencies in the regulatory T and NK
cell populations and negative hematopoietic cytokines (22,23).
These subsequently induce excessive apoptosis of CD34+
HSCs and inhibit hematopoietic colony formation through the
perforin, granzyme B, Fas/FasL and the TNF-related
apoptosis-inducing ligand pathways (13). Furthermore, an in-depth study of its
pathogenic mechanism suggested that both the numbers and function
of mDCs were significantly increased in the peripheral blood from
individuals with SAA (5,6,24).
mDCs secrete IL-12, a major stimulator, which induces the
differentiation of naive CD4+ T cells into the Th1
phenotype (25). Th1 cells were
reported to be overly activated subsequent to the excessive
secretion of cytokines such as IFN-γ and IL-2, which promote
CD8+ CTLs in patients with SAA (26). Thus, it may be inferred that the
pathogenesis of SAA is associated with improved function and
increased number of mDCs.
Although the understanding of the immune
pathogenesis of SAA gradually improved over numerous years of
research, the specific antigens, mDCs and T cells involved remained
unclear. Previous studies by our group have elucidated that the
immune cascade activation of SAA is closely relevant to the
upregulation and hyperfunction of mDCs, which may be the initiating
factor resulting in the activation of subsequent immune responses.
Liu et al (6) detected
changes in protein components of BM mDCs in the SAA group by
two-dimensional electrophoresis analysis. Compared with normal
controls, the expression of LILRA3 in mDCs of patients with SAA
increased. Coincidentally, Qi et al (7) also reported on the alterations in
protein expression levels of BM CD34+ cells in the SAA
group determined by iTRAQ analysis. Compared with that in healthy
participants, the expression of LILRA5 in CD34+ cells of
patients with SAA also increased.
LILRs are a family of immune-modulatory proteins
that are localized on human chromosome 19 in the region
19q13.4(9), principally expressed
on NK, myeloid, T and B cells. Receptors on LILRs modulate the
maturation of DCs and affect the antigen that presents DC
functions, thus regulating T-cell proliferation. LILRs are
progressively known as the critical regulators of innate immune
responses that act through the threshold modulation and amplitude
of lymphoid and myelomonocytic cell activation (27). LILRA3 is a soluble molecule
belonging to a household of highly homologous cell surface
receptors, principally expressed through mono-myeloid cells
(9). LILRA3 is unique as such that
it lacks cytoplasmic and transmembrane domains, and thus, is an
exclusively secreted protein. MHC class I molecules and Nogo 66 are
candidates for high-affinity binding with LILRA3 ligands. The
functions of LILRA3 remain to be fully elucidated. However, studies
suggested that it may act as a significant function in the
pathogenesis of autoimmune disorders. Serum LILRA3 protein levels
are significantly increased and are one of the strongest
independent markers of disease severity in multiple sclerosis.
Serum IL-10 and IFN-γ are positively correlated with LILRA3 levels
and negatively associated with serum TNF-α and LILRA3 in patients
with multiple sclerosis (12).
Serum LILRA3 concentrations are also significantly upregulated in
patients with rheumatoid arthritis (28), systemic lupus erythematosus
(29) and Sjögren's syndrome
(30), and positively correlated
with disease activity and severity. In the present study, the
expression of LILRAs in patients with SAA was investigated. LILRA3
mRNA expression was increased in BMMNCs of patients with SAA,
whereas others (LILRA1, -2, -4 and -6) were not significantly
different. Compared with healthy controls, the results verified
that the percentage of intracytoplasmic LILRA3+ on
CD11C+HLA-DR+ mDCs, the relative intensity of
LILRA3 protein in mDCs and the expression of LILRA3 mRNA in mDCs
were markedly enhanced in patients with SAA. Simultaneously, the
percentages of intracytoplasmic LILRA3+ in
CD34+ cells were also augmented, which positively
correlated with its expression in mDCs. These results suggested
that LILRA3 was abnormally present upstream of SAA in mDCs and that
there was an abnormal expression of downstream targets in
CD34+ cells. The present results suggested that the
expression of LILRA3 was increased in patients with SAA and that
this was positively correlated with disease progression, activity
and response to therapy.
The expression of LILRA3 mRNA was higher in patients
with new SAA and remission SAA in comparison with the control
group, but there were no differences between new SAA and remission
SAA. Although the expression of LILRA3 in remission SAA was not
significantly different from that of untreated SAA, the expression
of LILRA3 on mDCs of patients with remission SAA was lower than
that in the untreated SAA group. The reason may be that compared
with the normal control group, the cytokine storm period in the
remission SAA group was not over, but it was smaller than that in
the patients with untreated SAA. No statistically significant
difference in the expression of LILRA3 was obtained between SAA
patients with short-term remission (≤12 months) and long-term
remission (>12 months). A significant risk of relapse was
reported with rapid tapering of cyclosporine in patients with
remission SAA and the treatment should be continued for a long time
until the immunoreaction is completely back to normal (20). LILRA3 upregulated the transcription
of IL-1A, IL-1B and IL-6 and modulated the expression of
co-stimulatory molecules and MHC in B-cells and monocytes.
Signaling via LILRs affects the maturation and activation of DCs,
maintains the antigen-presenting properties of DCs and regulates
immune responses (27,30,31).
Thus, it may be possible that LILRA3 is involved in the
CTLs-mediated autoimmune pathogenesis of SAA and antagonizing
LILRA3 may be a novel strategy for addressing disease prevention
and progression. However, the present results provide insight into
several hypotheses about the pathogenesis of SAA. Future studies
delineating the exact mechanisms of LILRA3 in the pathogenesis of
SAA are warranted.
In conclusion, the present results suggested that
LILRA3 was highly expressed and positively correlated with disease
progression in the mDCs and CD34+ cells of patients with
SAA. Taken together, the present results indicate that LILRA3 is an
important participant in the pathogenesis of SAA and a potential
therapeutic target.
Acknowledgements
Not applicable.
Funding
Funding: This work was supported by the National Natural Science
Foundation of China (grant nos. 81970116, 81970115, 81870101 and
81500101) and the Tianjin Municipal Natural Science Foundation
(grant nos. 18JCYBJC91700 and 18ZXDBSY00140).
Availability of data and materials
Not applicable.
Authors' contributions
RF and ZS designed the study and revised the
manuscript. HY, HL and YZ performed experiments, analyzed data and
wrote the initial draft of the manuscript. HW, WQ, CL, ZL, SG, YS
and JT contributed to the experiments and the collection of
patients' features. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The study was in compliance with the Declaration of
Helsinki and was approved by the Ethics Committee of Tianjin
Medical University General Hospital (Tianjin, China). Written
informed consent was obtained from all participants.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Young NS: Aplastic anemia. N Engl J Med.
379:1643–1656. 2018.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Young NS: The etiology of acquired
aplastic anemia. Rev Clin Exp Hematol. 4:236–259. 2000.
|
|
3
|
Miano M and Dufour C: The diagnosis and
treatment of aplastic anemia: A review. Int J Hematol. 101:527–535.
2015.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Zeng Y and Katsanis E: The complex
pathophysiology of acquired aplastic anaemia. Clin Exp Immunol.
180:361–370. 2015.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Zonghong S, Meifeng T, Huaquan W, Limin X,
Jun W, Rong F, Hong L and Yuhong W: Circulating myeloid dendritic
cells are increased in individuals with severe aplastic anemia. Int
J Hematol. 93:156–162. 2011.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Liu C, Sheng W, Fu R, Wang H, Li L, Liu H
and Shao Z: Differential expression of the proteome of myeloid
dendritic cells in severe aplastic anemia. Cell Immunol.
285:141–148. 2013.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Qi W, Fu R, Wang H, Liu C, Ren Y, Shao Y
and Shao Z: Comparative proteomic analysis of CD34(+) cells in bone
marrow between severe aplastic anemia and normal control. Cell
Immunol. 304-305:9–15. 2016.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Norman PJ, Carey BS, Stephens HAF and
Vaughan RW: DNA sequence variation and molecular genotyping of
natural killer leukocyte immunoglobulin-like receptor, LILRA3.
Immunogenetics. 55:165–171. 2003.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Brown D, Trowsdale J and Allen R: The LILR
family: Modulators of innate and adaptive immune pathways in health
and disease. Tissue Antigens. 64:215–225. 2004.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Hirayasu K and Arase H: Functional and
genetic diversity of leukocyte immunoglobulin-like receptor and
implication for disease associations. J Hum Genet. 60:703–708.
2015.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Jones DC, Kosmoliaptsis V, Apps R, Lapaque
N, Smith I, Kono A, Chang C, Boyle LH, Taylor CJ, Trowsdale J and
Allen RL: HLA class I allelic sequence and conformation regulate
leukocyte Ig-like receptor binding. J Immunol. 186:2990–2997.
2011.PubMed/NCBI View Article : Google Scholar
|
|
12
|
An H, Lim C, Guillemin GJ, Vollmer-Conna
U, Rawlinson W, Bryant K and Tedla N: Serum leukocyte
immunoglobulin-like receptor A3 (LILRA3) is increased in patients
with multiple sclerosis and is a strong independent indicator of
disease severity; 6.7kbp LILRA3 gene deletion is not associated
with diseases susceptibility. PLoS One. 11(e0149200)2016.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Low HZ, Ahrenstorf G, Pommerenke C,
Habermann N, Schughart K, Ordóñez D, Stripecke R, Wilk E and Witte
T: TLR8 regulation of LILRA3 in monocytes is abrogated in human
immunodeficiency virus infection and correlates to CD4 counts and
virus loads. Retrovirology. 13(15)2016.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Sugahara-Tobinai A, Inui M, Metoki T,
Watanabe Y, Onuma R, Takai T and Kumaki S: Augmented ILT3/LILRB4
expression of peripheral blood antibody secreting cells in the
acute phase of kawasaki disease. Pediatr Infect Dis J. 38:431–438.
2019.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Debebe BJ, Boelen L and Lee JC: IAVI
Protocol C Investigators. Sanders EJ, Anzala O, Kamali A, Kaleebu
P, Karita E, Kilembe W, et al: Identifying the immune interactions
underlying HLA class I disease associations. Elife.
9(e54558)2020.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Camitta BM, Storb R and Thomas ED:
Aplastic anemia (second of two parts): Pathogenesis, diagnosis,
treatment, and prognosis. N Engl J Med. 306:712–718.
1982.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Camitta BM, Storb R and Thomas ED:
Aplastic anemia (first of two parts): Pathogenesis, diagnosis,
treatment, and prognosis. N Engl J Med. 306:645–652.
1982.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Di Bona E, Rodeghiero F, Bruno B, Gabbas
A, Foa P, Locasciulli A, Rosanelli C, Camba L, Saracco P, Lippi A,
et al: Rabbit antithymocyte globulin (r-ATG) plus cyclosporine and
granulocyte colony stimulating factor is an effective treatment for
aplastic anaemia patients unresponsive to a first course of
intensive immunosuppressive therapy. Gruppo italiano trapianto di
midollo osseo (GITMO). Br J Haematol. 107:330–334. 1999.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Method. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Liu C and Shao Z: Aplastic anemia in
China. J Transl Int Med. 6:134–137. 2018.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Liu C, Sun Y and Shao Z: Current concepts
of the pathogenesis of aplastic anemia. Curr Pharm Des. 25:236–241.
2019.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Chen T, Zhang T, Liu C, Wang C, Ding S,
Shao Z and Fu R: NK cells suppress CD8+ T cell immunity
via NKG2D in severe aplastic anemia. Cell Immunol. 335:6–14.
2019.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Liu B, Shao Y, Liu Z, Liu C, Zhang T and
Fu R: Bone marrow plasma cytokine signature profiles in severe
aplastic anemia. Biomed Res Int. 2020(8789275)2020.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Liu C, Zheng M, Wang T, Jiang H, Fu R,
Wang H, Ding K, Zhou Q and Shao Z: PKM2 is required to activate
myeloid dendritic cells from patients with severe aplastic anemia.
Oxid Med Cell Longev. 2018(1364165)2018.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Steinbrink K, Mahnke K, Grabbe S, Enk AH
and Jonuleit H: Myeloid dendritic cell: From sentinel of immunity
to key player of peripheral tolerance. Hum Immunol. 70:289–293.
2009.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Qi W, Yan L, Liu C, Fu R, Wang H and Shao
Z: Abnormal histone acetylation of CD8+ T cells in
patients with severe aplastic anemia. Int J Hematol. 104:540–547.
2016.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Park JE, Brand DD, Rosloniec EF, Yi AK,
Stuart JM, Kang AH and Myers LK: Leukocyte-associated
immunoglobulin-like receptor 1 inhibits T-cell signaling by
decreasing protein phosphorylation in the T-cell signaling pathway.
J Biol Chem. 295:2239–2247. 2020.PubMed/NCBI View Article : Google Scholar
|
|
28
|
An H, Chandra V, Piraino B, Borges L,
Geczy C, McNeil HP, Bryant K and Tedla N: Soluble LILRA3, a
potential natural antiinflammatory protein, is increased in
patients with rheumatoid arthritis and is tightly regulated by
interleukin 10, tumor necrosis factor-alpha, and interferon-gamma.
J Rheumatol. 37:1596–1606. 2010.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Du Y, Sun F, Zhou M, Wu X, Sun W, Jiang Y,
Cheng Q, Chen X, Wu H and Xue J: The expression and clinical
significance of different forms of LILRA3 in systemic lupus
erythematosus. Clin Rheumatol. 38:3099–3107. 2019.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Low HZ, Reuter S, Topperwien M,
Dankenbrink N, Peest D, Kabalak G, Stripecke R, Schmidt RE,
Matthias T and Witte T: Association of the LILRA3 deletion with
B-NHL and functional characterization of the immunostimulatory
molecule. PLoS One. 8(e81360)2013.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Nezos A, Evangelopoulos ME and Mavragani
CP: Genetic contributors and soluble mediators in prediction of
autoimmune comorbidity. J Autoimmun. 104(102317)2019.PubMed/NCBI View Article : Google Scholar
|