Introduction
Hemophagocytic lymphohistiocytosis (HLH), also known
as hemophagocytic syndrome, is an abnormal immunoregulation
syndrome characterized by excessive immune activation and
hyperinflammatory disorder (1). HLH
is generally divided into two types: Primary or familial HLH and
secondary HLH (2). Primary or
familial HLH results from genetic mutations, while secondary HLH
occurs in infections, autoimmune diseases, malignancies,
immunodeficiencies and organ transplantations (2,3). The
global incidence of HLH in children is 1-1.2 per 100,000 with a
median age of onset of 1.8 years (4-7).
Mortality in HLH ranges from 20-75% according to different reports,
including children and adults (3,6). After
the International Histiocyte Society HLH-2004 treatment protocol
was widely adopted, the 5-year survival rate of patients with this
condition increased from 54 to 66% (8). According to these standards (2), identification of ≥5 of the following
indicators can be used to confirm HLH diagnosis: i) Fever; ii)
splenomegaly; iii) cytopenia (affecting two or three lineages;
hemoglobin <90 g/l; platelets <100x109/l;
neutrophil absolute value <1.0x109/l); iv)
hypertriglyceridemia (≥3 mmol/l) and/or hypofibrinogenemia (≤1.5
g/l); v) serum ferritin increase (>500 ug/l); vi) plasma soluble
CD25 [soluble interleukin (IL)-2 receptor] increase (≥2,400 U/ml);
vii) decrease or absence in natural killer (NK) cell activity; and
viii) hemophagocytosis in the bone marrow, spleen or lymph nodes
with no evidence of malignancy.
The onset of HLH is usually acute and the clinical
manifestations of this critical illness are associated with
pro-inflammatory cytokine storms and macrophage infiltration into
tissue (9,10). Cytotoxicity of impaired NK cells is
the hallmark of HLH pathogenesis (11). Simultaneously, severe immune
dysfunction contributes to excessive proliferation and persistent
activation of antigen-presenting cells, uncontrolled cytokine
secretion and proliferation, and ectopic migration of macrophages
and T cells (12). Moreover, the
high levels of pro-inflammatory cytokines, such as tumor necrosis
factor (TNF)-α, IL-6, and interferon (IFN)-γ, suppress
hematopoiesis and lipoprotein lipase levels, resulting in cytopenia
and hyperlipidemia (9,10). These biological events are
considered to be the causes of multiple organ failure and the high
mortality of HLH (13).
Acute kidney injury (AKI) is a common complication
of HLH. The global incidence of AKI concurrent with HLH varies from
8-62%, with 33.3-59.0% of patients with AKI requiring renal
replacement (14,15). Thrombotic microangiopathy (TMA) is a
rare disease characterized by microthrombus formation which
obstructs the vascular cavity. Primary TMA can be induced by
thrombotic thrombocytopenic purpura (TTP) and atypical hemolytic
uremic syndrome (aHUS). Secondary TMA is often associated with the
Shiga toxin, due to Escherichia coli, and pregnancy-related
TMAs (16-18).
However, TMA associated with AKI in combination with HLH is rarely
reported. To the best of our knowledge, no reports have been
published regarding infants, among whom this disease remains
intractable (3). Due to the lack of
characteristic clinical manifestations, providing a clear diagnosis
of HLH remains challenging. The present study presents a case of
HLH with TMA in an infant who showed a favorable prognosis after
experiencing complications in diagnosis and treatment.
Case report
Patient information
A 15-month-old male presenting with a weeklong fever
and reduced platelet and hemoglobin levels was admitted to Anhui
Provincial Children's Hospital (Hefei, China) in November 2018. No
improvement was observed after two days of antibiotic and antiviral
treatment. No significant past medical history or family history
was recorded.
Clinical findings
On admission, the axillary temperature of the
patient was 41˚C, and the duration of the fever was prolonged. The
patient's parents stated that the fever was accompanied by a chill
and a facial rash, which subsided when the fever retreated.
Physical examination revealed pharyngeal congestion and
paleness.
Diagnostic assessment
Hematology test
The main features of the hematology test on the
first day after admission indicated pancytopenia (hemoglobin, 9.3
g/dl; platelet count, 79x109/l; neutrophil count,
2.79x109/l), severe inflammation (C-reactive protein,
50.91 mg/l; procalcitonin, 12.16 ng/l), AKI (serum creatinine, 188
µmol/l) and proteinuria (2+). Whole blood cell count decreased
progressively (hemoglobin, 7.5 g/dl; platelet count,
26x109/l; neutrophil count, 1.1x109/l).
Concurrently, the causes of persistent fever and AKI
remained unclear with the following tests reporting negative
results: Hemoculture, Widal reaction, rheumatoid factor,
antinuclear antibody series, antineutrophil cytoplasmic antibody,
hepatitis A, hepatitis B, hepatitis C virus, syphilis, HIV,
Legionella immunoglobulin M (IgM), mycoplasma IgM, chlamydia
IgM, rickettsia IgM, adenovirus IgM, respiratory and cytotoxic
virus IgM, influenza A/B virus IgM and parainfluenza virus IgM. The
patient tested positive for the IgG antibodies of Epstein-Barr
virus (EBV), cytomegalovirus (CMV) and the rubella virus, but the
IgM antibody results were negative. In addition, tests for the
presence of nucleic acids of EBV and CMV were negative. These
findings suggested that the patient had previously contracted EBV,
CMA and the rubella virus, but that these viruses were not active.
Complement C3 and C4 were in the normal range (based on the
observation in our hospital (The First Affiliated Hospital of Anhui
Medical University, Hefei, China), C3 0.87-1.41 g/l, C4 0.1-0.4
g/l). A blood smear did not detect any malarial parasites and the
percentage of broken red blood cells was 1.3% (detailed information
is described in Table SI).
Imaging examination
Low dose chest CT produced normal results and
echocardiography did not show coronary artery dilation. At the
acute phase, abdominal ultrasound suggested a diffuse enlargement
of both kidneys (right kidney 104x33 mm, left kidney 100x32 mm),
while the size of the liver and spleen were normal (data not
shown).
Genetic testing
The strategy of genetic testing was whole-exome
sequencing (WES) and the report showed there were two
lysosomal-trafficking regulator (LYST) variants. The sequencing was
performed by Beijing Chigene Translational Medicine Research Center
Co., Ltd. The protocol of WES was as follows: i) Sample collection.
The EDTA-treated peripheral blood was collected with informed
consent of the patient's parents; ii) DNA extraction. The fetal
tissue genomic DNA was extracted using a Blood Genome Column Medium
Extraction kit (CW2298; CoWin Biosciences, Inc.) according to the
manufacturer's instructions. The extracted DNA samples were
subjected to quality control using a Qubit 2.0 fluorimeter and
electrophoresis with 0.8% agarose gel; iii) whole exome library
construction. Protein-coding exome enrichment was performed using
xGen Exome Research Panel v1.0 (Integrated DNA Technologies, Inc.)
that consisted of 429,826 individually synthesized and
quality-controlled probes, which targeted a 39 Mb protein-coding
region (19,396 genes) of the human genome and covered 51 Mb of
end-to-end tiled probe space; and iv) sequencing. High-throughput
sequencing was performed using an Illumina NovaSeq 6000 series
sequencer (PE150; Illumina, Inc.) and ≥99% of the target sequence
was sequenced. The next step was bioinformatics analysis: i)
Quality control. Raw data were processed by fastp for adapter
removal and low-quality read filtering; ii) Variant calling. The
paired-end reads were performed using Burrows-Wheeler Aligner (BWA,
version 0.7.11) to the Ensembl GRCh37/hg19 reference genome. Base
quality score recalibration together with SNP and short indel
calling was conducted using the Genome Analysis Toolkit. According
to the sequence depth and variant quality, SNPs and indels were
screened to ensure that high quality and reliable variants were
obtained; iii) Variant annotation and pathogenicity prediction. The
online system independently developed by Chigene (www.chigene.org; 12/21/2018) was used to annotate both
database-based minor allele frequencies (MAFs) and the American
College of Medical Genetics and Genomics practice guideline-based
pathogenicity of every yielded gene variant (19). The system also provided serial
software packages for conservative analysis and protein product
structure prediction.
Therapeutic intervention
Initially, ceftriaxone and ganciclovir were
administered due to the persistent fever. On the second day after
admission, AKI was diagnosed by hematological parameters according
to the Kidney Disease Improving Global Outcomes (KDIGO) AKI
criteria of 2012(20) of an
increase in serum creatinine (Scr) within 48 h >26.5 µmol/l or
Scr within 7 days up to 1.5 times that of the base value.
Indicators for infection, such as C-reactive protein and
procalcitonin levels, increased progressively. Therefore,
hemodialysis was initiated and antibiotics were upgraded to
meropenem and teicoplanin. Human serum albumin, intravenous
immunoglobulin (IVIG), plasma and suspended red blood cell
transfusion were administered to stabilize hemodynamics and
low-dose dexamethasone (0.1-0.3 mg/kg/day) was used as an
anti-inflammatory and for suppression of the immune response.
Follow-up and outcomes
Serum ferritin levels increased progressively, from
950 to 2,237 µg/l within 6 days and serum IL-2 receptor levels were
>7,500 U/ml (normal range based on the observation in our
hospital (The First Affiliated Hospital of Anhui Medical
University, Hefei, China), 223-710 U/ml). The infant's fibrinogen
levels sharply decreased from 4.27 g/l on the third day of
admission to 1.66 g/l on the 26th day (normal range based on the
observation in our hospital (The First Affiliated Hospital of Anhui
Medical University, Hefei, China), 2-4 g/l). Triglyceridemia
increased progressively, from 1.75 to 5.07 mmol/l. Moreover, NK
cell count was initially normal, but decreased in the convalescent
phase. Concurrently, the number of reticulocytes initially appeared
to reduce but increased during the recovery period. Urine protein
was between 2+ and 3+ throughout the period.
Imaging examination
During the recovery period, an abdominal ultrasound
before discharge showed that the kidneys were in a recovery period
from diffuse renal disease (right kidney 90x32 mm, left kidney
82x33 mm; data not shown).
To establish the pathogenesis of the fever, a bone
marrow biopsy was performed on the sixth day of admission. Informed
consent for this procedure was obtained from the patient's parents.
A syringe was used to apply suction to the needle until marrow
appeared. Tissue preparation: i) Prepare a film: A drop of bone
marrow was placed onto one end of the smear slide. The spreader
slide was placed at an angle of approximately 30-40° in
front of the drop and slid backwards until it came into contact
with the sample drop. The spreader slide was advanced forwards,
generating a smear with a feathered edge. ii) Smear staining: The
smear was first dipped into methanol to fix the specimens and
subsequently placed in Wright's-Giemsa stain (Beijing Solarbio
Science & Technology Co., Ltd.) for 10-15 min for staining at
room temperature (20-25˚C). The smear was next moved to a mixture
of Wright's-Giemsa stain and pH 6.8 phosphate buffer for 5 min at
room temperature (20-25˚C). After the staining step, the smear was
given a quick rinse in distilled water and was allowed to dry
before cover-slipping. iii) Microscopy detection: Olympus BX53
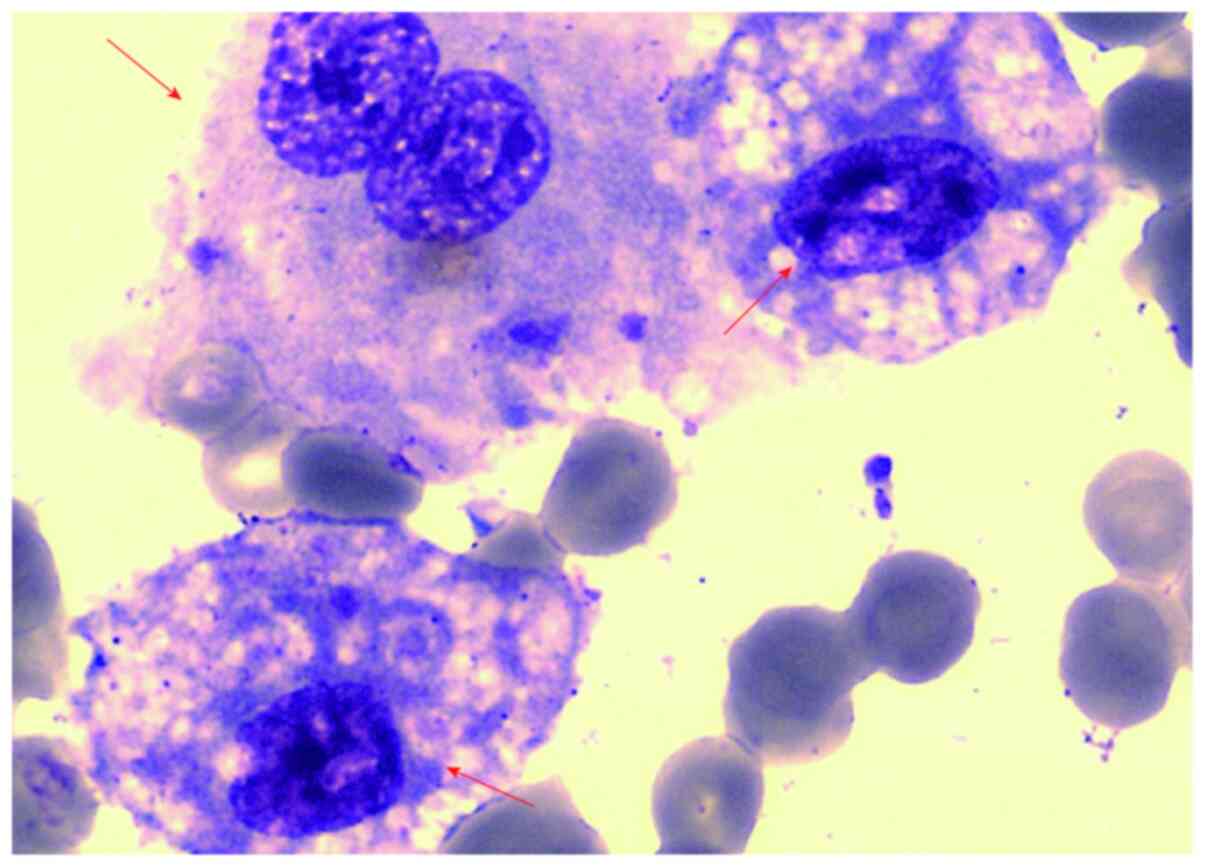
(Olympus Corporation), Magnification, x100. HLH was considered due
to the presence of mononuclear and phagocytic tissue cells in the
bone marrow (Fig. 1). However, the
recovery of renal function was not obvious after hemodialysis. To
further clarify the etiology, a renal biopsy was performed on the
11th day of admission. Kidney tissue Preparation: Tissue for light
microscopy (LM) was needed to keep the specimen in the vial of 10%
formalin (Sigma-Aldrich; Merck KGaA) for 12-24 h. Regarding
electron microscopy, the cortex with glomeruli (cut 1-2 mm from
each end of the core) was placed in the vial that contained 2.5%
glutaraldehyde solution (Sigma-Aldrich; Merck KGaA). Kidney tissue
sectioning: The specimen was processed through dehydration,
clearing, paraffin infiltration, embedding and then sectioning the
paraffin blocks at a thickness of 2-3 µm. Kidney tissue staining:
For light microscopy: Hematoxylin and eosin (H&E) staining kit
(Beyotime Institute of Biotechnology) and Masson stain kit (Abcam)
were used for the staining. Image examination: Light microscope
x400 (Olympus Corporation; BX53), scanning electron microscope x700
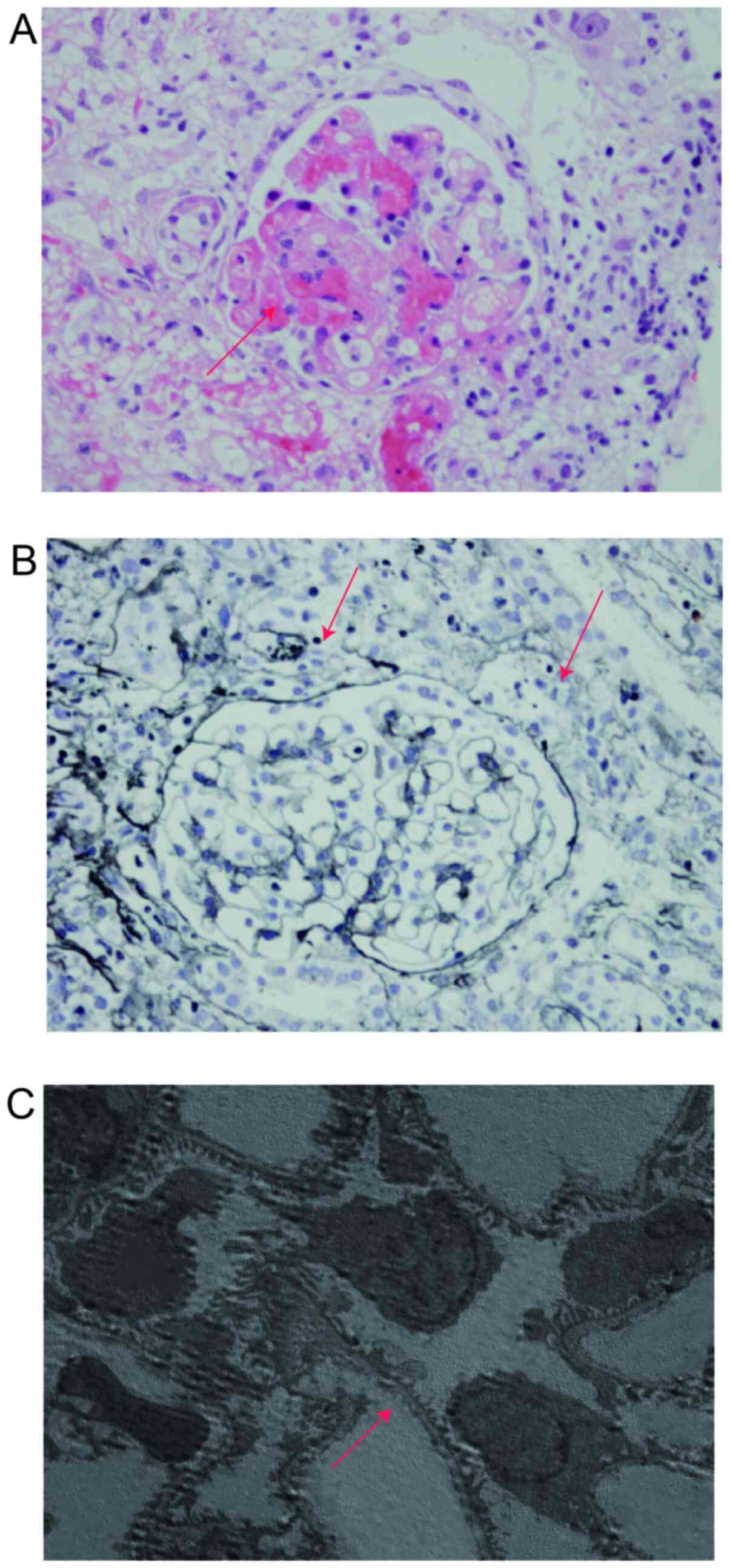
(Hitachi, Ltd.). The glomeruli presented with diffuse minor
lesions, a high number of microthrombus formations (5/11) and
moderate tubulointerstitial lesions (Fig. 2). At this point, the child met the
HLH diagnostic criteria and was consequently diagnosed with HLH
complicated with TMA kidney damage.
Following HLH diagnosis, dexamethasone was
administered according to the 2004 diagnostic and therapeutic
guidelines for HLH (2). Due to the
patient's age, immunosuppressive agents were not used. On the 14th
day after admission, the patient's body temperature returned to
normal and hemoglobin and platelet parameters were stable. On the
21st day of admission, serum creatinine levels was gradually
decreased and the dialysis catheter was removed after six cycles of
hemodialysis. The results of the auxiliary examination and
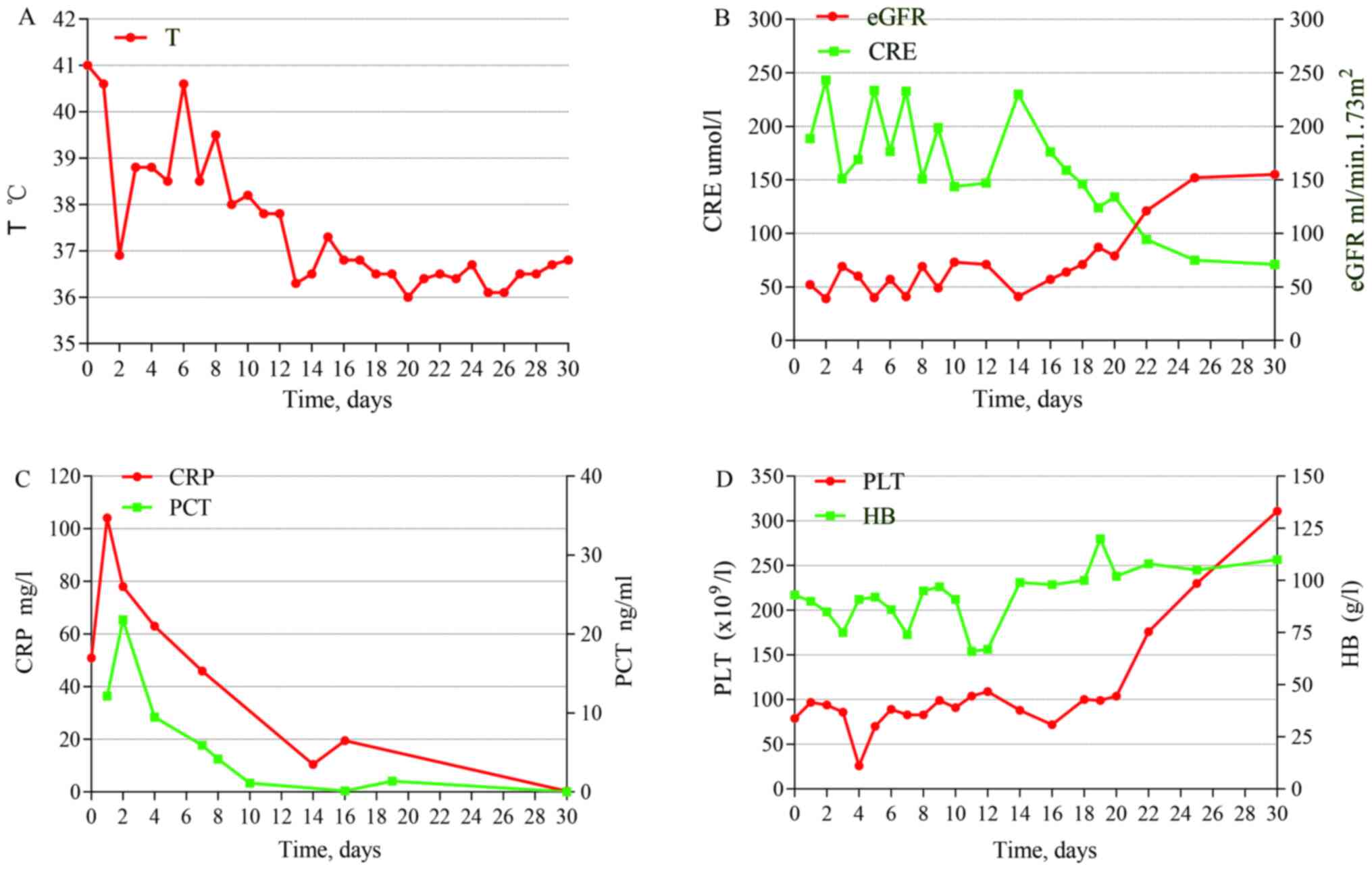
treatment are shown in Fig. 3.
The cause of HLH in the patient remained unclear.
Thus, genetic testing was performed. Within the whole-exome
sequencing detection range, there was a variation in the LYST gene.
However, no disease-related mutations were detected (Fig. 4). A follow-up examination after 18
months showed no sign of HLH recurrence and that serum creatinine
levels were in the normal range.
Discussion
In the present case report, an infant was admitted
to Anhui Provincial Children's Hospital with a persistent fever for
7 days. Laboratory tests showed anemia, thrombocytopenia and AKI.
Initially, sepsis combined with multiple organ dysfunction was
diagnosed, but detailed examinations did not support the diagnosis.
Following renal biopsy and bone marrow aspiration, the diagnosis
was clearly shown to be HLH. Although the clinical presentations of
the patient were atypical at first, the infant met six indicators
of the HLH-2004 diagnostic criteria (2) on the 12th day after admission.
Patients may not have all these clinical features on initial
presentation and features may be accompanied by other symptoms,
making diagnosis challenging (3).
In the present case, the initial increase in
fibrinogen may be associated with the hypercoagulable state of the
child, and fibrinogen levels decreased in the recovery stage due to
the hypercoagulable improvement. Moreover, the reduction of
reticulocytes at the acute phase and the rise during the recovery
period may be associated with myelosuppression. In the present
case, no spleen enlargement was noted during the course of
treatment, which differed from the majority of HLH cases (2).
In the present case, rheumatism and tumor were
excluded through the results of the examinations. Echocardiography
did not show coronary artery dilation, and the patient did not
present with Kawasaki-like symptoms. Moreover, although ganciclovir
can induce AKI (21), the patient
presented with renal dysfunction prior to ganciclovir treatment.
AKI caused by prerenal and postrenal factors was excluded, as the
patient did not show any signs of volume depletion and urinary
tract obstruction. Atypical hemolytic uremic syndrome (aHUS) has
many similar clinical features with HLH accompanied with AKI and
TMA. According to the latest classification criteria presented at
the KDIGO Controversies Conference in 2016(22), >50% of patients with aHUS have an
underlying inherited and/or acquired complement abnormality, which
results in dysregulated activity of the alternative pathway at the
endothelial cell surface. In the present case, aHUS was not
considered to be the diagnosis, since the complement C3 and C4 of
the patient were in the normal range, the number of reticulocytes
were reduced and the broken red blood cells in the peripheral blood
were normal. The prognosis for aHUS is poor, as the disease will
advance to end-stage renal disease within 2 years of presentation
in the majority of patients who do not receive effective treatment
such as eculizumab (22). During
the 18-month follow-up, the renal function of the patient was
normal, which indicated that the diagnosis of HLH with AKI and TMA
was correct.
The accepted treatment protocol for HLH is based on
2004 guidelines (2), in which
dexamethasone is recommended as the main therapy, while etoposide
(VP16), methotrexate and cyclosporine A are major components of the
treatment regimen (23).
Additionally, hematopoietic stem cell transplantation (HSCT) is
recognized as a recovery treatment (24). The main treatments administered in
the present case report were dexamethasone and specific supporting
therapy. Immunosuppressive agents were not used due to the
patient's age. Supporting therapy, including dialysis and IVIG were
administered and the clinical signs of the infant progressively
improved. The present treatment protocol was in line with previous
reports (25-28),
in which immunosuppressive agents were not used and the outcome was
also satisfying. Haytoglu et al (29) reported that patients with secondary
HLH were treated with corticosteroids, IVIG and physical
examination without chemotherapeutics and that hospital survival
rate was 100%. Therefore, the present case report presents evidence
for the potential benefit of these therapeutic protocols, in which
glucocorticoids (dexamethasone in this case) is the first-line drug
and IVIG is recommended to all patients with HLH. As for the use of
immunosuppressive agents, all clinical conditions should be taken
into account and integrated and individualized treatments are
hypothesized to be beneficial to patients.
In order to search the existing clinical cases of
HLH with TMA, ‘hemophagocytic lymphohistiocytosis’ and ‘renal
thrombotic microangiopathy’ were used as key terms when searching
the PubMed database (https://pubmed.ncbi.nlm.nih.gov/). Clinical features
of HLH with TMA in previously reported cases are summarized in
Table I. Aside from the present
case report, only one other case involving a child was reported,
and the male to female ratio was 1:1. One case was infected with
CMV, one case was infected with EBV, two cases were organ
transplant patients, one was infected with parvovirus B19 and four
cases were idiopathic. AKI was present in 7/8 cases. Proteinuria
was present in 5/8 cases. Dialysis was required for three patients
and plasma exchange was required for one patient. Glucocorticoids
and IVIG were administered in all patients. Immunosuppressive
agents were only applied in two patients. Seven patients recovered
and one patient succumbed to the disease.
 | Table IFeatures of cases of hemophagocytic
lymphohistiocytosis with thrombotic microangiopathy reported in the
literature. |
Table I
Features of cases of hemophagocytic
lymphohistiocytosis with thrombotic microangiopathy reported in the
literature.
| Characteristic
patient reference no. | Value |
|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
|---|
| Age, years | 63 | 18 | 24 | 18 | 60 | 39 | 15 | 1 |
| Sex | Female | Female | Male | Female | Female | Male | Male | Male |
| Etiology | CMV | Idiopathic | Transplant
Parvovirus B19 | Idiopathic | Idiopathic | Transplant | EBV | Idiopathic |
| Renal biopsy
result |
|
Light
microscopy | TMA | TMA | TMA | TMA | TMA | TMA | TMA | TMA |
|
Immuno-fluorescence | Negative | Negative | Negative | Negative | Negative | NA | IgM | Negative |
|
Electron
microscopy | NA | Fluffy
material | NA | Histiocytic
infiltration | Foot process
fusion | NA | Foot process
fusion | Macrophage
infiltration |
| AKI | Yes | Yes | Yes | Yes | Yes | Yes | No | Yes |
| Dialysis | HD | No | No | No | HD | PE | No | HD |
| Treatment |
|
Steroids | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
|
IVIG | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
|
Immunosuppressive
agents | No | No | No | No | Yes | Yes | No | No |
| Outcome | Death | Cure | Cure | Cure | Cure | Cure | Cure | Cure |
| Author, year | Thaunat et
al, 2006 | Thaunat et
al, 2006 | Ardalan et
al, 2008 | Chiang et
al, 2002 | Bae et al,
2016 | Esmaili et
al, 2015 | XX Chu et
al, 2017 | Present case |
| (Ref.) | (25) | (25) | (26) | (27) | (30) | (31) | (28) | Present case |
One Japanese study reported that Mucor
infection was associated with TMA in HLH (32). There were three cases of
clinically-diagnosed HLH complicated TMA which did not undergo
biopsy (33,34). Bae et al (30) reported that TMA in HLH may occur
with greater frequency than previously known, since diagnostic
biopsies have rarely been performed in patients with HLH. If AKI
occurred in patients with HLH, it is recommended that clinicians
should also observe whether TMA is present in the patients as
well.
The present case report hypothesized that the cause
of the AKI was renal TMA, as the renal biopsy showed red blood cell
debris obstructing the glomerular capillary lumen, severe tubular
focal atrophy and microthrombosis in the arterioles. TMA is a
histopathological lesion of the arteriole and capillary thickening,
including intraluminal platelet thrombosis and vessel obstruction
(17). In TMA pathogenesis,
endothelial injury is a key factor in the initiation of microemboli
formation (35,36). The mononuclear-macrophage system is
activated under the action of cytokines, releasing free radicals
and proteolytic enzymes, resulting in endothelial cell damage and
inflammatory reactions, thereby triggering TMA (30,33).
Similarly, systemically-activated lymphocytes and macrophages and
overproduction of highly elevated proinflammatory cytokines in the
circulation such as IFN, TNF, IL-6 and macrophage-colony
stimulating factor are pathogenetic characteristics of HLH
(6,37). The immune dysfunction in TMA and in
HLH has some overlap in pathophysiology, and clinicians should be
aware of TMA when patients experience AKI with HLH.
Infections, malignancy and autoimmune disorders are
the main causes of the secondary HLH (13). EBV is the most reported infection
(3,38), and several studies have reported
that infections of CMV, herpes simplex viruses 1 and 2,
varicella-zoster virus, roseolovirus, Kaposi's sarcoma-associated
herpesvirus and Parvovirus B19 have been detected in secondary HLH
(13,25,26,39).
However, the present case report involved blood EB virus
antibodies, nucleic acid detection and renal tissue EB virus
antibody detection, all of which were negative. Moreover, the
present case report also excluded EBV infection, malignancy and
autoimmune disorders.
Primary HLH is generally combined with clear
familial inheritance or genetic problems, where it most often
presents in the first year of life as an autosomal recessive
disorder due to homozygous mutations (40). The annual incidence of primary HLH
in Sweden is 0.12-0.15 per 100,000 children per year (41). There are different types of familial
HLH, including familial HLH types 2-5, X-linked lymphoproliferative
disorder type 1, Griscelli syndrome type 2, Chediak-Higashi
syndrome, X-linked lymphoproliferative disorder type 2, mutation of
the Nod-like receptor family, caspase recruitment domain-containing
4, lysinuric protein intolerance and the mutation of heme oxygenase
1, whereby the associated genes were PRF1, UNC13D, STX11, STXBP2,
SH2D1A, RAB27A, LYST, BIRC4, NLRC4, SLC7A7, HMOX1(13). LYST gene mutation may lead to
pigment abnormalities due to abnormal melanocyte granules (42). It is worth noting that there is a
variation in the LYST gene in the present patient, which can cause
Chediak-Higashi syndrome (13), but
most variations in the LYST gene are harmless to primary HLH
(13) and there was no evidence
demonstrating the association between LYST and this disease. The
association between the LYST gene and hemophagocytic syndrome is
still ambiguous, so at present, the etiology of HLH in the patient
is not clear. Further analyses have demonstrated that ~15% of
patients with adult HLH harbor mutations in familial HLH genes that
may serve as predisposing alleles for the development of HLH,
though still in response to typical triggers (6).
Few large multicenter research studies have
addressed the prognosis for children with HLH worldwide. A previous
study (6) found that the survival
rate of patients with HLH was only 10% before
immunochemotherapeutic treatment strategies were administered, and
once the HLH-94 guidelines were applied, the five-year overall
survival rate was 54%. Nandhakumar et al (3) revealed that 77% (35/45) of children
with secondary HLH recovered completely; however, all 13 patients
with primary HLH died, despite the therapies administered according
to the HLH 2004 protocol. To date, primary HLH has lacked effective
treatment, and HSCT is recognized as the only viable treatment
option to physicians. The latest clinical trial demonstrated that
emapalumab is an efficacious targeted therapy for patients <18
years with primary HLH (43).
The present case report suggests that clinicians
should consider TMA in patients with HLH with AKI. The major
limitation of the present case report is the unknown pathogenesis
of the disease. Since the genetic testing was negative and the
follow-up inquiry into the child revealed that he had recovered,
the cause of HLH was suspected to be an infection. However, the
specific pathogenic microorganism causing this was not found,
therefore, the present case is likely to be one of idiopathic
HLH.
Supplementary Material
Blood biochemical indices and
treatment protocol
Acknowledgements
The authors would like to thank Mr. Zhiming Zheng
(Chigene Translational Medicine Research Center Co., Ltd., Beijing,
China), who performed whole exome sequencing for the patient.
Funding
Funding: The present study was supported by grants from the
Anhui Province Public Welfare Technology Application Research
Project (CN; grant no. 1704f0804027) and the Excellent Young
Talents Fund Program of Higher Education Institutions of Anhui
Province (grant no. gxbjZD07).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HYG performed the kidney biopsy, analysed the
clinical data, and wrote the manuscript. JMS performed the
histological examination and stated the presented diagnosis. YS
interpreted the patient data and revised the manuscript. QZ and XX
performed the kidney biopsy, performed the histological
examination, and interpreted the patient data. RH analyzed and
interpreted the patient’s clinical data and contributed to writing
the manuscript. DF analyzed and interpreted the patient data and
revised the final manuscript. All authors read and approved the
final manuscript..
Ethics approval and consent to
participate
All procedures performed in the study were in
accordance with the Ministry of Health ethics review on biomedical
research involving human subjects, The declaration of Helsinki and
the Council for International Organizations of Medical Sciences
International ethical guidelines for biomedical research. Written
informed consent was obtained from the family of the patient
described in this report.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Skinner J, Yankey B and Shelton BK:
Hemophagocytic lymphohistiocytosis. Adv Crit Care. 30:151–164.
2019.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Henter JI, Horne A, Aricó M, Egeler RM,
Filipovich AH, Imashuku S, Ladisch S, McClain K, Webb D, Winiarski
J and Janka G: HLH-2004: Diagnostic and therapeutic guidelines for
hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer.
48:124–131. 2007.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Nandhakumar D, Loganatha A, Sivasankaran
M, Sivabalan S and Munirathnam D: Hemophagocytic
lymphohistiocytosis in children. Indian J Pediatr. 87:526–531.
2020.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Erker C, Harker-Murray P and Talano JA:
Usual and unusual manifestations of familial hemophagocytic
lymphohistiocytosis and langerhans cell histiocytosis. Pediatr Clin
North Am. 64:91–109. 2017.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Allyson Niece M, Zora R..Rogers M,
Naveed-Ahmad M, Anne-Marie-Langevin M and McClain KL:
Hemophagocytic lymphohistiocytosis in Texas: Observations on
ethnicity and race. Pediatr Blood Cancer. 54:424–428.
2010.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Allen CE and McClain KL: Pathophysiology
and epidemiology of hemophagocytic lymphohistiocytosis. Hematology
Am Soc Hematol Educ Program. 2015:177–182. 2015.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Henter J, Elinder G, Söder O and Ost A:
Incidence in Sweden and clinical features of familial
hemophagocytic lymphohistiocytosis. Acta Paediatr Scand.
80:428–435. 1991.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Bergsten E, Horne AC, Aricó M, Astigarraga
I, Egeler RM, Filipovich AH, Ishii E, Janka G, Ladisch S, Lehmberg
K, et al: Confirmed efficacy of etoposide and dexamethasone in HLH
treatment: Long-term results of the cooperative HLH-2004 study.
Blood. 130:2728–2738. 2017.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Frimmel S, Hinz M, Schipper J, Bogdanow S,
Mitzner S and Koball S: Cytokine adsorption is a promising tool in
the therapy of hemophagocytic lymphohistiocytosis. Int J Artif
Organs. 42:658–664. 2019.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Zinter MS and Hermiston ML: Calming the
storm in HLH. Blood. 134:103–104. 2019.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Egeler RM, Shapiro R, Loechelt B and
Filipovich A: Characteristic immune abnormalities in hemophagocytic
lymphohistiocytosis. J Pediatr Hematol Oncol. 18:340–345.
1996.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Usmani GN, Woda BA and Newburger PE:
Advances in understanding the pathogenesis of HLH. Br J Haematol.
161:609–622. 2013.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Al-Samkari H and Berliner N:
Hemophagocytic lymphohistiocytosis. Annu Rev Pathol. 13:27–49.
2018.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Aulagnon F, Lapidus N, Canet E, Galicier
L, Boutboul D, Peraldi MN, Reuter D, Bernard R, Schlemmer B,
Azoulay E and Zafrani L: Acute kidney injury in adults with
hemophagocytic lymphohistiocytosis. Am J Kidney Dis. 65:851–859.
2015.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Karapinar B, Yilmaz D, Balkan C, Akin M,
Ay Y and Kvakli K: An unusual cause of multiple organ dysfunction
syndrome in the pediatric intensive care unit: Hemophagocytic
lymphohistiocytosis. Pediatr Crit Care Med. 10:285–290.
2009.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Bayer G, Von Tokarski F, Thoreau B,
Bauvois A, Barbet C, Cloarec S, Mérieau E, Lachot S, Garot D,
Bernard L, et al: Etiology and outcomes of thrombotic
microangiopathies. Clin J Am Soc Nephrol. 14:557–566.
2019.PubMed/NCBI View Article : Google Scholar
|
|
17
|
George JN and Nester CM: Syndromes of
thrombotic microangiopathy. N Engl J Med. 371:654–666.
2014.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Barbour T, Johnson S, Cohney S and Hughes
P: Thrombotic microangiopathy and associated renal disorders.
Nephrol Dial Transplant. 27:2673–2685. 2012.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American College
of Medical Genetics and Genomics and the Association for Molecular
Pathology. Genet Med. 17:405–424. 2015.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Kidney Disease: Improving Global Outcomes:
KDIGO Clinical Practice Huideline for Acute Kidney Injury. KDIGO
AKI Guideline. Online Appendices A-F:2012.
|
|
21
|
Dos Santos Mde F, Dos Santos OF, Boim MA,
Razvickas CV, De Moura LA, Ajzen H and Schor N: Nephrotoxicity of
acyclovir and ganciclovir in rats: Evaluation of glomerular
hemodynamics. J Am Soc Nephrol. 8:361–367. 1997.PubMed/NCBI
|
|
22
|
Goodship TH, Cook HT, Fakhouri F, Fervenza
FC, Frémeaux-Bacchi V, Kavanagh D, Nester CM, Noris M, Pickering
MC, Rodríguez de Córdoba S, et al: Atypical hemolytic uremic
syndrome and C3 glomerulopathy: Conclusions from a ‘Kidney Disease:
Improving Global Outcomes’ (KDIGO) controversies conference. Kidney
Int. 91:539–551. 2017.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Sadaat M and Jang S: Hemophagocytic
lymphohistiocytosis with immunotherapy: Brief review and case
report. J Immunother Cancer. 6(49)2018.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Schram AM and Berliner N: How I treat
hemophagocytic lymphohistiocytosis in the adult patient. Blood.
125:2908–2914. 2015.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Thaunat O, Delahousse M, Fakhouri F,
Martinez F, Stephan JL, Noël LH and Karras A: Nephrotic syndrome
associated with hemophagocytic syndrome. Kidney Int. 69:1892–1898.
2006.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Ardalan MR, Shoja MM, Tubbs RS, Esmaili H
and Keyvani H: Postrenal transplant hemophagocytic
lymphohistiocytosis and thrombotic microangiopathy associated with
parvovirus B19 infection. Am J Transplant. 8:1340–1344.
2008.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Chiang WC, Wu MS, Tsai CC, Lin SL, Tsai TJ
and Hsieh BS: Thrombotic microangiopathy in hemophagocytic
syndrome: A case report. J Formos Med Assoc. 101:362–367.
2002.PubMed/NCBI
|
|
28
|
XX Chu, Zhang Y and Liao SJ: A case of HLH
complicated with renal damage of thrombotic microangiopathy. Chin J
Nephrol. 33:625–626. 2017.
|
|
29
|
Haytoglu Z, Yazici N and Erbay A:
Secondary hemophagocytic lymphohistiocytosis: Do we really need
chemotherapeutics for all patients? J Pediatr Hematol Oncol.
39(e106-e109)2017.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Bae MN, Kwak DH, Park SJ, Choi BS, Park
CW, Choi YJ, Lee JW, Yang CW, Kim YS and Chung BH: Acute kidney
injury induced by thrombotic microangiopathy in a patient with
hemophagocytic lymphohistiocytosis. BMC Nephrol.
17(4)2016.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Esmaili H, Mostafidi E, Mehramuz B,
Ardalan M and Mohajel-Shoja M: An update on renal involvement in
hemophagocytic syndrome (macrophage activation syndrome). J
Nephropathol. 5:8–14. 2016.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Inagaki N, Sugimoto K, Hosone M, Isobe Y,
Yamamoto Y, Sasaki M, Kato A, Mori T and Oshimi K: Disseminated
Mucor infection and thrombotic microangiopathy in
lymphoma-associated hemophagocytic syndrome. Int J Hematol.
88:355–356. 2008.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Ardalan MR: Review of thrombotic
microangiopathy (TMA), and post-renal transplant TMA. Saudi J
Kidney Dis Transpl. 17:235–244. 2006.PubMed/NCBI
|
|
34
|
Ishiguro T, Kojima A, Shimizu T, Mita N,
Kuroiwa S and Takayanagi N: Combined hemophagocytic syndrome and
thrombotic microangiopathy due to mixed infection with influenza
virus and pneumococcal pneumonia. Clin Case Rep. 7:131–134.
2019.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Yu XJ, Yu F, Song D, Wang SX, Song Y, Liu
G and Zhao MH: Clinical and renal biopsy findings predicting
outcome in renal thrombotic microangiopathy: A large cohort study
from a single institute in China. ScientificWorldJournal.
2014(680502)2014.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Goldberg RJ, Nakagawa T, Johnson RJ and
Thurman JM: The role of endothelial cell injury in thrombotic
microangiopathy. Am J Kidney Dis. 56:1168–1174. 2010.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Canna SW and Marsh RA: Pediatric
hemophagocytic lymphohistiocytosis. Blood. 135:1332–1343.
2020.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Mărginean MO, Molnar E and Chinceşan MI:
Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis in
a small child: A case report. Medicine (Baltimore).
99(e18759)2020.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Munoz J, Shareef N and Donthireddy V:
Cytomegalovirus-induced haemophagocytic lymphohistiocytosis
syndrome. BMJ Case Rep. 2012(bcr1020114963)2012.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Esteban YM, de Jong JLO and Tesher MS: An
overview of hemophagocytic lymphohistiocytosis. Pediatr Ann.
46:e309-e313:2017.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Meeths M, Horne A, Sabel M, Bryceson YT
and Henter JI: Incidence and clinical presentation of primary
hemophagocytic lymphohistiocytosis in Sweden. Pediatr Blood Cancer.
62:346–352. 2014.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Risma KA and Marsh RA: Hemophagocytic
lymphohistiocytosis: Clinical presentations and diagnosis. J
Allergy Clin Immunol Pract. 7:824–832. 2019.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Locatelli F, Jordan MB, Allen C, Cesaro S,
Rizzari C, Rao A, Degar B, Garrington TP, Sevilla J, Putti MC, et
al: Emapalumab in children with primary hemophagocytic
lymphohistiocytosis. N Engl J Med. 382:1811–1822. 2020.PubMed/NCBI View Article : Google Scholar
|