Introduction
Liver cancer is the sixth common cancer and the
second leading cause of death from cancer worldwide, which is
frequently diagnosed at late stage and characterized by rapid
progression (1,2). Patients with advanced liver cancer
are ineligible for surgical resection and other potentially
curative treatments (3). In
addition, treatment efficacy for liver cancer is poor, due to its
resistance to conventional chemotherapy (4,5).
After decades of searching for effective therapeutic agents for
liver cancer, systemic treatment with sorafenib has been
established as the standard therapy for advanced liver cancer
(6). Furthermore, regorafenib has
been demonstrated to have a high efficacy and safety in patients
who experience liver cancer progression during sorafenib treatment
(7). Regorafenib, a bi-aryl urea
compound, is an antiangiogenic and antitumorigenic agent approved
for the treatment of patients with advanced liver cancer during
sorafenib therapy; it inhibits tumor growth by targeting multiple
kinases, including vascular endothelial growth factor receptors
1-3, platelet-derived growth factor receptor β, c-KIT, RET, B-RAF,
fibroblast growth factor receptor 1 and serine/threonine kinase
(Raf and p38 MAPK) (8).
Regorafenib has been revealed to suppress cell proliferation,
invasion and angiogenesis via the ERK/NF-κB signaling pathway
(9). However, patients with
advanced liver cancer develop resistance to regorafenib via the
activation of the AKT signaling pathway, and the overall outcome of
patients with advanced liver cancer is far from satisfactory
(10). Therefore, the development
of more effective therapeutic agents and strategies for liver
cancer is required.
Harmine, a naturally occurring β-carboline, is
isolated from a medicinal herb traditionally used in the Middle
East and North Africa, known as Peganum harmala L. (11). It has a wide range of
pharmacological activities, including anti-microbial, anti-fungal,
anti-oxidative and anticancer activities (12). Furthermore, harmine is an ideal
drug candidate for liver cancer therapy, as it is selectively
harmful to liver cancer cells but has minimal side effects on
normal liver cells (13). Harmine
possesses notable anticancer properties by targeting apoptosis,
autophagy, abnormal cell proliferation, angiogenesis and metastasis
(14). Harmine displays
pharmacological activities by suppressing substrate phosphorylation
in the dual-specificity tyrosine-regulated kinase (DYRK) family,
and it exhibits the highest affinity for DYRK1A (15). Furthermore, combining harmine with
other agents, such as Bcl-2 inhibitors and osimertinib, has been
identified as a potential approach to overcoming resistance to
chemotherapy (16,17). In the present study, it was
examined whether harmine combined with regorafenib may be a
potential therapeutic regimen for liver cancer treatment.
Materials and methods
Materials
Harmine (cat. no. HY-N0737A) was purchased from
MedChemExpress and regorafenib (cat. no. A8236) was obtained by
APeXBIO Technology LLC. The primary antibodies against
cleaved-caspase-3 (cat. no. 9661S), cleaved-poly (ADP-ribose)
polymerase (cleaved-PARP; cat. no. 9541S; 1:1,000) and DYRK1A (cat.
no. 2771S; 1:1,000) were obtained from Cell Signaling Technology,
Inc. The primary antibodies against PARP-1/2 (H-250) (cat. no.
sc-7150; 1:500), anti-GAPDH antibody (FL-335) (cat. no. sc-25778;
1:500), myeloid cell leukemia-1 (Mcl-1) (S-19) (cat. no. sc-819;
1:500), phosphorylated (p)-AKT (1/2/3) (Ser473) (cat. no. sc-7985;
1:500) and caspase-3 (H-277) (cat. no. sc-7148; 1:500) were
purchased from Santa Cruz Biotechnology, Inc. The primary antibody
against α-tubulin (rabbit polyclonal antibody; cat. no. AF0001;
1:1,000) was obtained from Beyotime Institute of Biotechnology. The
primary antibody against AKT (cat. no. 610836; 1:500) was purchased
from BD Biosciences. The secondary antibodies, including
DyLight™ 800 4X PEG-conjugated anti-rabbit-IgG (cat. no.
5151) and DyLight™ 800 4X PEG-conjugated anti-mouse-IgG
(cat. no. 5470), were obtained from Cell Signaling Technology,
Inc.
Cell culture
All cell lines were obtained from the Shanghai
Institute of Biochemistry and Cell Biology. HepG2 cells were
maintained in DMEM (cat. no. C0006; Hangzhou Keyi Shengwu Jishu
Youxian Gongsi) with 10% FBS (cat. no. P30-3302; PAN-Biotech GmbH)
and Hep3B cells were cultured in MEM (cat. no. C0032, Hangzhou
KEYI) with 10% FBS and 100 U/ml penicillin/streptomycin (Gibco;
Thermo Fisher Scientific, Inc.). Mycoplasma testing was performed
on these cell lines, which were then determined and authenticated
for genotypes using short tandem repeat DNA fingerprinting and
passaged for <6 months (18).
Liver cancer cells were divided into four groups: control group
treated with DMSO, harmine treated group, regorafenib treated
group, harmine plus regorafenib group.
Cytotoxicity assay
The proliferation of HepG2 and Hep3B cells was
measured using sulforhodamine blue (SRB) cytotoxicity assay. The
cells were inoculated into a 96-well plate at a density of
8x103 cells/well. When the cell confluence reached 30%,
harmine, regorafenib, harmine plus regorafenib were added into the
plates. After 72 h, cells were fixed with 10% trichloroacetic acid
for 8 h at 4°C. Next, tap water was used to wash the
96-well plates. After the wells had dried up, 0.4% SRB (cat. no.
230162-5G; Sigma-Aldrich; Merck KGaA) solution was used to stain
the cells 30 min at room temperature. Next, the unbound dye was
removed using 1% acetic acid washing in the 96-well plates.
Tris-based solution (10 mM) was used to solubilize the SRB dye.
Finally, cell proliferation was detected as previously described
using a microplate reader at 570 nm (19).
Detection of cell death
Cell death was detected using PI (cat. no. ST511;
Beyotime Institute of Biotechnology) followed by flow cytometry
detection as previously described (20). HepG2 and Hep3B cells were
inoculated at a density of 2x104 per well into a 96-well
plate and incubated with harmine (4 µM) and/or regorafenib (1 µM or
2 µM) for 48 h at 37°C. The collected cells were then
fixed and permeabilized with 75% precooled ethanol at 4˚C for 2 h.
Next, 400 µl PBS containing 50 µg/ml RNase A (cat. no. ST579;
Beyotime Institute of Biotechnology) was used to treat cells at
37˚C for 30 min. Subsequently, cells were incubated with 5 µl PI
solution for 15 min at room temperature and analyzed using a
FACSCalibur cytometer (FACSCalibur; BD Biosciences). Finally, the
sub-G1 peak was analyzed by Cellquest Pro Software (version 6.0; BD
Biosciences), and gating was performed to keep cell death <10%
in the control group.
Colony formation assay
HepG2 and Hep3B cells were inoculated into a 6-cm
dish at a low cell density (6x103 cells per dish) to
evaluate the ability of cells to form colonies. Then, the cells
were incubated with 1 µM harmine, 2 µM regorafenib and harmine plus
regorafenib for 14 days at 37°C. The sensitivity of drug
treatment is dependent on cell density, drug treatment was
indicated to achieve maximal anti-proliferative effect at low cell
density and retain the anti-proliferative effect at intermediate
cell density (data not shown). Thus, the concentrations of harmine
and regorafenib used in colony formation assay were different
compared with that in apoptotic detection. Dishes were fixed with
10% paraformaldehyde solution for 30 min at room temperature and
stained with 1% crystal violet solution for 30 min at room
temperature, and colonies containing >50 cells were counted
using ImageJ software (National Institutes of Health; version
v1.8.0). The survival fractions were calculated according to the
following equation: Survival fractions=cell number of treated
sample/cell number of control (21).
Western blot analysis
HepG2 and Hep3B cells were incubated with
harmine/regorafenib for 48 h at 37°C. Cells were
incubated with lysis buffer (cat. no. P0013, Beyotime Institute of
Biotechnology) on ice for 30 min. The concentrations of protein
were determined by the BCA method using an Enhanced BCA Protein
Assay kit (cat. no. P0009; Beyotime Institute of Biotechnology).
Proteins (20 µg/lane) were fractionated on 8-12% Tris-glycine gels,
and following electrophoresis, proteins were transferred to PVDF
membranes. The membranes were blocked with 5% skimmed milk for 2 h
at room temperature and then washed three times with 0.1%
TBS-Tween-20 (TBST), followed by incubation with the primary
antibodies (cleaved-caspase-3, cleaved-PARP, DYRK1A, PARP-1/2,
GAPDH, Mcl-1, p-AKT (Ser473), caspase-3, α-tubulin and AKT)
overnight. Next, membranes were washed three times with TBST,
incubated and visualized with DyLight™ 800 4X
PEG-conjugated anti-rabbit-IgG (cat. no. 5151) and
DyLight™ 800 4X PEG-conjugated anti-mouse-IgG (cat. no.
5257) (1:50,000; Cell Signaling Technology, Inc.) secondary
antibodies for 2 h, washed three times again with TBST and scanned
using an imaging system (Odyssey CLX Image Studio; version 5.0.21;
LiCor Odyssey CLx imager; LI-COR Biosciences).
RNA interference
HepG2 and Hep3B cells were seeded in six-well plates
(2x105 cells/well) and then transfected with 40 nM
DYRK1A small interfering (si)RNA using jetPRIME
(Polyplus-transfection SA) at 37˚C. After 24 h siRNA transfection,
HepG2 and Hep3B cells were collected and seeded on 96-well plates
(4x103 cells/well) at 37˚C overnight. Cells were then
treated with regorafenib (0.5, 1, 2, 4 and 8 µM) for 72 h, and cell
proliferation was detected using an SRB assay, as described above.
For the detection of western blotting, after 24 h siRNA
transfection, HepG2 and Hep3B cells were collected and incubated
with lysis buffer, followed by western blot analysis. DYRK1A siRNAs
were synthetized by Shanghai GenePharma Co., Ltd. The siRNA
sequences used were as follows: siDYRK1A-1,
5'-AUGGAGCUAUGGACGUUAADTDT-3'; siDYRK1A-2,
5'-AAACUCGAAUUCAACCUUADTDT-3'; and negative control,
5'-UUCUCCGAACGUGUCACGUDTDT-3'.
Plasmid transfection
HepG2 cells reached 80% confluence in a 6-cm plate
prior to transfection. Attractene transfection reagent was
purchased from Qiagen AB. Constitutively active AKT1 (CA-AKT; cat.
no. 78778; pcDNA3.1-HA AKT1) plasmid was obtained from Addgene,
Inc. CA-AKT or pcDNA3.1 plasmid (cat. no. V87020; Invitrogen;
Thermo Fisher Scientific, Inc.) transfection was performed using
Attractene transfection reagent with 1 µg plasmid per reaction at
37˚C for 24 h, according to the manufacturer's protocol (22). Cells were then treated with 4 µM
harmine + 1 µM regorafenib for 48 h, after which the expression of
the indicated proteins were detected.
Statistical analysis
All data are presented as the mean ± SD. All
experiments were conducted at least three times, and representative
results are presented. A two-tailed unpaired Student's t-test was
used to determine the differences between two groups. One-way ANOVA
followed by Tukey's post hoc test was used to examine the
significant differences among multiple groups using GraphPad
(Version 6.01; GraphPad Software). The combination index (CI)
values were calculated using CalcuSyn (Version 2.0; Biosoft;
synergism, CI<0.9; additive effect, 0.9-1.10; antagonism,
>1.10). P<0.05 was considered to indicate a statistically
significant difference.
Results
DYRK1A knockdown reinforces the
anticancer effects of regorafenib in vitro
DYRK1A serves a vital role in drug sensitivity in
cancer treatment (17,16). In the present study, two DYRK1A
siRNA significantly suppressed the expression of DYRK1A and DYRK1A
knockdown by siDYRK1A-1 reinforced the anticancer effects of
regorafenib in liver cancer cells by suppressing cell proliferation
(Fig. 1A and B).
Harmine plus regorafenib
synergistically inhibits the proliferation of liver cancer
cells
As expected, the DYRK1A inhibitor harmine plus
regorafenib significantly suppressed the proliferation of liver
cancer cells compared with single-agent treatment (Fig. 2A). To verify the synergistic
anti-liver cancer effect of harmine plus regorafenib, CI values
were calculated. As revealed in Fig.
2B, co-treatment with harmine and regorafenib exhibited
synergistic anti-proliferative effects on liver cancer cells
(CI<0.7). Particularly in HepG2 cells, harmine plus regorafenib
exhibited a strong synergy (CI<0.1). In addition, harmine could
increase the suppression of colony formation by regorafenib in
liver cancer cells (Fig. 3). Thus,
these data revealed that harmine enhanced the anticancer effects of
regorafenib by inhibiting liver cancer cell proliferation.
Harmine increases regorafenib-induced
cell death
To further confirm the synergistic anti-liver cancer
effect of harmine plus regorafenib, PI staining was used to detect
cell death. As demonstrated in Fig.
4A, the cell death proportion of HepG2 cells (sub-G1) was 3.20%
in the control group, 9.98% in the regorafenib group, 49.81% in the
harmine group and 80.40% in the combination treatment group.
Therefore, harmine markedly enhanced regorafenib-induced cell death
in HepG2 cells (Fig. 4B). The
enhanced cell death induced by harmine plus regorafenib was also
observed in Hep3B cells, compared with single agent treatment.
These data revealed that harmine plus regorafenib significantly
induced cell death in liver cancer cells.
Harmine combined with regorafenib
induces cell death via the AKT pathway
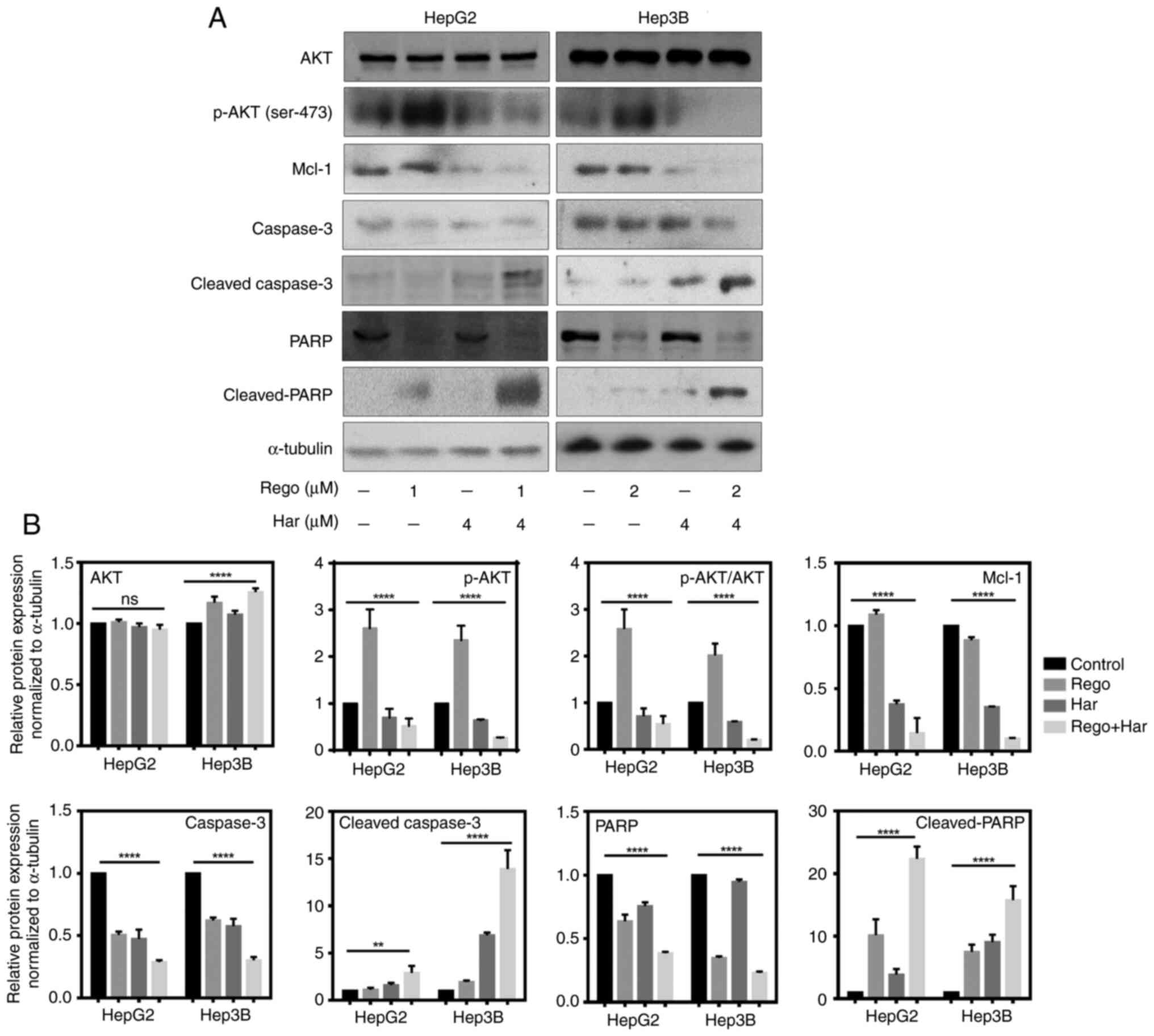
Furthermore, western blot analysis detecting
cleaved-PARP and cleaved-caspase-3 confirmed that harmine plus
regorafenib treatment promoted apoptosis in liver cancer cells when
compared with single agent treatment (Fig. 5A and B). The phosphorylation of AKT was
enhanced in liver cancer cells treated with regorafenib compared
with the DMSO treated group, while harmine plus regorafenib
significantly suppressed p-AKT compared with single agent treatment
in HepG2 and Hep3B cells. Furthermore, harmine plus regorafenib
significantly inhibited the expression of Mcl-1 compared with
single agent treatment.
Overexpression of AKT reverses the
apoptosis induced by regorafenib plus harmine in liver cancer
cells
To determine the role of the AKT signaling pathway
in regorafenib plus harmine treatment, AKT was overexpressed by
transfecting the CA-AKT plasmid into liver cancer cells.
Regorafenib plus harmine induced apoptosis in the
AKT-overexpression group was lower than that in empty vector group,
indicating that AKT overexpression reduced the apoptosis induced by
regorafenib plus harmine in liver cancer cells (Fig. 6A and B). Thus, these results demonstrated that
AKT signaling may serve a vital role in regorafenib plus harmine
treatment.
Discussion
The activation of the PI3K/AKT/mTOR pathway is
involved in the development and proliferation of liver cancer stem
cells during acquired sorafenib resistance (23). Although regorafenib is the only
systemic therapy demonstrated to provide survival advantages in
patients with liver cancer experiencing disease progression on
sorafenib treatment, the activation of the AKT pathway still
restricts the anticancer activity of regorafenib during acquired
regorafenib resistance (24,25).
This highlighted the need for alternative therapeutic strategies
for targeting the PI3K/AKT/mTOR pathway during
sorafenib/regorafenib treatment. Mcl-1 is vital to cancer treatment
due to its upregulation in a wide variety of human cancer types,
including non-small-cell lung cancer, breast cancer, ovarian
cancer, prostate cancer and pancreatic cancer (26,27).
The PI3K/AKT/mTOR pathway could increase the stabilization of the
Mcl-1 protein by inhibiting Mcl-1 phosphorylation in human melanoma
(28). It has been reported that
DYRK1A overexpression has no effect on hepatic PI3K/AKT activation
in mouse liver (29). However, the
AKT pathway is activated by DYRK1A in the brain of mice with
hyperhomocysteinemia, and treatment with harmine has been
demonstrated to diminish AKT activation by reducing AKT
phosphorylation (30).
Furthermore, trophinin associated protein has been indicated to
directly bind to DYRK1A or DYRK1B, leading to the cytoplasmic
retention of DYRK1A or DYRK1B and inducing cell cycle progression
via AKT activation (31).
Therefore, it was suggested that AKT may be abnormally activated by
DYRK1A during the pathogenesis of multiple diseases or development
of drug resistance. The results of the present study indicated that
AKT was activated in regorafenib-treated liver cancer cells, and
that DYRK1A inhibition by harmine could suppress p-AKT and
reinforce the anti-liver cancer activity of regorafenib by
inhibiting the AKT pathway. These data highlighted that harmine may
be a compound that could reverse regorafenib resistance during
liver cancer treatment. However, the effect of harmine plus
regorafenib treatment on normal liver cells may require further
investigation.
Liver cancer is closely associated with fibrosis and
chronic inflammation arising from different etiologies, including
alcoholic and non-alcoholic fatty liver disease and hepatitis B and
C (32). Furthermore, 80-90% of
patients with liver cancer have underlying cirrhosis caused by
chronic liver inflammation (33).
Liver cancer develops in an intricate microenvironment
characterized by chronic inflammation (34). Thus, effective therapeutic
approaches for liver cancer are expected to prevent chronic
inflammation and oncogene-activated liver cancer growth (35). DYRK1A serves a critical role in
regulating the balance between T helper 17 and T regulatory (Treg)
cells, thereby contributing to the progression of inflammatory
disease, and harmine attenuates inflammation by regulating Treg
cell differentiation (36).
Furthermore, DYRK1A suppression has been indicated to destabilize
EGFR and reduce EGFR-dependent glioblastoma growth, and the
pharmacological inhibition of DYRK1A has been revealed to inhibit
stem cell behavior (37). These
findings highlight the potential importance of DYRK1A in liver
carcinogenesis and the need for the development of therapeutic
strategies to target DYRK1A in liver cancer treatment. In the
present study, to the best of our knowledge, it was reported for
the first time that DYRK1A inhibition may be an efficient way of
reinforcing the anticancer activity of regorafenib in liver cancer
treatment. However, preclinical studies in vivo and clinical
studies are required to verify the anti-liver cancer effect of
harmine plus regorafenib. In addition, the efficiency of DYRK1A
suppression plus regorafenib on inflammation during liver cancer
development requires further investigation.
In conclusion, DYRK1A knockdown increased the
anti-proliferative activity of regorafenib, and harmine enhanced
the effects of regorafenib in liver cancer cells by suppressing
cell proliferation and inducing apoptosis. Furthermore, harmine
suppressed the expression of p-AKT and enhanced the anticancer
activity of regorafenib via regulating the AKT pathway (Fig. 7). Thus, harmine may be a pertinent
sensitizer to regorafenib, and harmine plus regorafenib may be an
effective strategy for liver cancer treatment.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by Zhejiang Provincial
Natural Science Foundation of China (grant nos. LTY21H160001,
LY21H160017 and HDMY22H160421), Public-service Technology Research
Plan of Zhejiang Province (grant no. LGF21H310002), Scientific and
Technological Developing Scheme of Hangzhou City (grant no.
20191203B49), Zhejiang Provincial Medical and Health Technology
Project (grant nos. 2021433724 and 2020RC026) and National College
Student Innovation and Entrepreneurship Training Program (grant no.
202113021025).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
CZ and LZ designed the study. ZC, ZL, JL, SZ and JY
performed the experiments. JW performed the statistical analysis.
CZ wrote the manuscript. ZC and ZL confirmed the authenticity of
all the raw data. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Calderaro J, Ziol M, Paradis V and
Zucman-Rossi J: Molecular and histological correlations in liver
cancer. J Hepatol. 71:616–630. 2019.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Gomes MA, Priolli DG, Tralhão JG and
Botelho MF: Hepatocellular carcinoma: Epidemiology, biology,
diagnosis, and therapies. Rev Assoc Med Bras (1992). 59:514–524.
2013.PubMed/NCBI View Article : Google Scholar : (In English,
Portuguese).
|
|
3
|
Wu Q and Qin SK: Features and treatment
options of Chinese hepatocellular carcinoma. Chin Clin Oncol.
2(38)2013.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Avila MA, Berasain C, Sangro B and Prieto
J: New therapies for hepatocellular carcinoma. Oncogene.
25:3866–3884. 2006.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Fu J and Wang H: Precision diagnosis and
treatment of liver cancer in China. Cancer Lett. 412:283–288.
2018.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Zhu YJ, Zheng B, Wang HY and Chen L: New
knowledge of the mechanisms of sorafenib resistance in liver
cancer. Acta Pharmacol Sin. 38:614–622. 2017.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Fondevila F, Méndez-Blanco C,
Fernández-Palanca P, González-Gallego J and Mauriz JL: Anti-tumoral
activity of single and combined regorafenib treatments in
preclinical models of liver and gastrointestinal cancers. Exp Mol
Med. 51:1–15. 2019.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Wilhelm SM, Dumas J, Adnane L, Lynch M,
Carter CA, Schütz G, Thierauch KH and Zopf D: Regorafenib (BAY
73-4506): A new oral multikinase inhibitor of angiogenic, stromal
and oncogenic receptor tyrosine kinases with potent preclinical
antitumor activity. Int J Cancer. 129:245–255. 2011.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Chiang CH, Chung JG and Hsu FT:
Regorefenib induces extrinsic/intrinsic apoptosis and inhibits
MAPK/NF-κB-modulated tumor progression in bladder cancer in vitro
and in vivo. Environ Toxicol. 34:679–688. 2019.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Sun H, Feng F, Xie H, Li X, Jiang Q, Chai
Y, Wang Z, Yang R, Li R and Hou J: Quantitative examination of the
inhibitory activation of molecular targeting agents in
hepatocellular carcinoma patient-derived cell invasion via a novel
in vivo tumor model. Animal Model Exp Med. 2:259–268.
2019.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Li S, Wang A, Gu F, Wang Z, Tian C, Qian
Z, Tang L and Gu Y: Novel harmine derivatives for tumor targeted
therapy. Oncotarget. 6:8988–9001. 2015.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Prasad Kushwaha J, Baidya D and Patil S:
Harmine-loaded galactosylated pluronic F68-gelucire 44/14 mixed
micelles for liver targeting. Drug Dev Ind Pharm. 45:1361–1368.
2019.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Zhang L, Zhang F, Zhang W, Chen L, Gao N,
Men Y, Xu X and Jiang Y: Harmine suppresses homologous
recombination repair and inhibits proliferation of hepatoma cells.
Cancer Biol Ther. 16:1585–1592. 2015.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Jalali A, Dabaghian F and Zarshenas MM:
Alkaloids of Peganum harmala: Anticancer biomarkers with
promising outcomes. Curr Pharm Des. 27:185–196. 2021.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Seifert A, Allan LA and Clarke PR: DYRK1A
phosphorylates caspase 9 at an inhibitory site and is potently
inhibited in human cells by harmine. FEBS J. 275:6268–6280.
2008.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Li Y, Zhou D, Xu S, Rao M, Zhang Z, Wu L,
Zhang C and Lin N: DYRK1A suppression restrains Mcl-1 expression
and sensitizes NSCLC cells to Bcl-2 inhibitors. Cancer Biol Med.
17:387–400. 2020.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Li YL, Ding K, Hu X, Wu LW, Zhou DM, Rao
MJ, Lin NM and Zhang C: DYRK1A inhibition suppresses STAT3/EGFR/Met
signalling and sensitizes EGFR wild-type NSCLC cells to AZD9291. J
Cell Mol Med. 23:7427–7437. 2019.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Li YL, Zhang NY, Hu X, Chen JL, Rao MJ, Wu
LW, Li QY, Zhang B, Yan W and Zhang C: Evodiamine induces apoptosis
and promotes hepatocellular carcinoma cell death induced by
vorinostat via downregulating HIF-1α under hypoxia. Biochem Biophys
Res Commun. 498:481–486. 2018.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Vichai V and Kirtikara K: Sulforhodamine B
colorimetric assay for cytotoxicity screening. Nat Protoc.
1:1112–1116. 2006.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Riccardi C and Nicoletti I: Analysis of
apoptosis by propidium iodide staining and flow cytometry. Nat
Protoc. 1:1458–1461. 2006.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Zhang C, Shi J, Mao SY, Xu YS, Zhang D,
Feng LY, Zhang B, Yan YY, Wang SC, Pan JP, et al: Role of p38 MAPK
in enhanced human cancer cells killing by the combination of
aspirin and ABT-737. J Cell Mol Med. 19:408–417. 2015.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Zhang D, Yan B, Yu S, Zhang C, Wang B,
Wang Y, Wang J, Yuan Z, Zhang L and Pan J: Coenzyme Q10 inhibits
the aging of mesenchymal stem cells induced by D-galactose through
Akt/mTOR signaling. Oxid Med Cell Longev.
2015(867293)2015.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Kahraman DC, Kahraman T and Cetin-Atalay
R: Targeting PI3K/Akt/mTOR pathway identifies differential
expression and functional role of IL8 in liver cancer stem cell
enrichment. Mol Cancer Ther. 18:2146–2157. 2019.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Chen W, Yang J, Zhang Y, Cai H, Chen X and
Sun D: Regorafenib reverses HGF-induced sorafenib resistance by
inhibiting epithelial-mesenchymal transition in hepatocellular
carcinoma. FEBS Open Bio. 9:335–347. 2019.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Mirone G, Perna S, Shukla A and Marfe G:
Involvement of notch-1 in resistance to regorafenib in colon cancer
cells. J Cell Physiol. 231:1097–1105. 2016.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Xiang W, Yang CY and Bai L: MCL-1
inhibition in cancer treatment. Onco Targets Ther. 11:7301–7314.
2018.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Wei SH, Dong K, Lin F, Wang X, Li B, Shen
JJ, Zhang Q, Wang R and Zhang HZ: Inducing apoptosis and enhancing
chemosensitivity to gemcitabine via RNA interference targeting
Mcl-1 gene in pancreatic carcinoma cell. Cancer Chemother
Pharmacol. 62:1055–1064. 2008.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Jin L, Hu WL, Jiang CC, Wang JX, Han CC,
Chu P, Zhang LJ, Thorne RF, Wilmott J, Scolyer RA, et al:
MicroRNA-149*, a p53-responsive microRNA, functions as an oncogenic
regulator in human melanoma. Proc Natl Acad Sci USA.
108:15840–15845. 2011.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Noll C, Tlili A, Ripoll C, Mallet L, Paul
JL, Delabar JM and Janel N: Dyrk1a activates antioxidant NQO1
expression through an ERK1/2-Nrf2 dependent mechanism. Mol Genet
Metab. 105:484–488. 2012.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Abekhoukh S, Planque C, Ripoll C, Urbaniak
P, Paul JL, Delabar JM and Janel N: Dyrk1A, a serine/threonine
kinase, is involved in ERK and Akt activation in the brain of
hyperhomocysteinemic mice. Mol Neurobiol. 47:105–116.
2013.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Li L, Wei JR, Song Y, Fang S, Du Y, Li Z,
Zeng TT, Zhu YH, Li Y and Guan XY: TROAP switches DYRK1 activity to
drive hepatocellular carcinoma progression. Cell Death Dis.
12(125)2021.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Yang YM, Kim SY and Seki E: Inflammation
and liver cancer: Molecular mechanisms and therapeutic targets.
Semin Liver Dis. 39:26–42. 2019.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Ringelhan M, Pfister D, O'Connor T,
Pikarsky E and Heikenwalder M: The immunology of hepatocellular
carcinoma. Nat Immunol. 19:222–232. 2018.PubMed/NCBI View Article : Google Scholar
|
|
34
|
He M, Zhang W, Dong Y, Wang L, Fang T,
Tang W, Lv B, Chen G, Yang B, Huang P and Xia J: Pro-inflammation
NF-κB signaling triggers a positive feedback via enhancing
cholesterol accumulation in liver cancer cells. J Exp Clin Cancer
Res. 36(15)2017.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Koike K: Expression of junB is markedly
stimulated by glycyrrhizin in a human hepatoma cell line. Oncol
Rep. 25:609–617. 2011.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Khor B, Gagnon JD, Goel G, Roche MI,
Conway KL, Tran K, Aldrich LN, Sundberg TB, Paterson AM, Mordecai
S, et al: The kinase DYRK1A reciprocally regulates the
differentiation of Th17 and regulatory T cells. Elife.
4(e05920)2015.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Pozo N, Zahonero C, Fernández P, Liñares
JM, Ayuso A, Hagiwara M, Pérez A, Ricoy JR, Hernández-Laín A,
Sepúlveda JM and Sánchez-Gómez P: Inhibition of DYRK1A destabilizes
EGFR and reduces EGFR-dependent glioblastoma growth. J Clin Invest.
123:2475–2487. 2013.PubMed/NCBI View
Article : Google Scholar
|