Introduction
Vitamin D signaling plays a role in regulating blood
pressure through influencing vascular endothelial function,
oxidative stress and activation of the renin-angiotensin-system
(RAS), as well as increasing insulin resistance. The widespread
effect of vitamin D relies on the extensive presence of the vitamin
D receptor (VDR), which is expressed in every human tissue and
nearly all nucleated cells, although at varying levels. It is
currently hypothesized that almost all biological actions of
vitamin D are mediated by its active form, 1,25(OH)2D,
signaling mainly through the intracellular VDR (1). Vitamin D signaling has been
associated with elevated plasma renin and angiotensin (Ang) II
levels (2). Animal experiments
have indicated that 1,25(OH)2D3 can inhibit
renin gene transcription (3), and
Zhou et al (4) revealed
that blood pressure is higher in
1α(OH)ase-/- mice compared with
wild-type mice, which is accompanied by elevated mRNA
expression levels of renin, plasma aldosterone and Ang II. These
studies indicate that 1,25(OH)2D3 may
influence blood pressure by regulating the central and peripheral
RAS through an anti-oxidative stress mechanism. Another in
vivo experiment (5) suggested
that vitamin D deficiency increases Ang II and oxygen anion levels
in local vascular smooth muscle cells (VSMCs). Several studies have
also demonstrated that 1,25(OH)2D3 deficiency
can increase blood pressure by inducing oxidative stress pathways
and over-activating the central RAS (6-8).
When vitamin D levels are adequate, a number of the
intracellular oxidative stress-related activities are
downregulated. Suboptimal concentrations of serum 25(OH)D fail to
subdue oxidative stress conditions, augment intracellular oxidative
damage and decrease the rate of apoptosis. Superoxide dismutase
(SOD) belongs to a group of antioxidant enzymes that play a
significant role in regulating oxidative stress in cells (9). Peroxiredoxin 4(Prdx4), a typical
endoplasmic reticulum-resident 2-Cys antioxidant of peroxiredoxins,
can fine-tune hydrogen peroxide catabolism, which affects cell
survival by affecting redox balance, oxidative protein folding and
hydrogen peroxide signaling (10).
Vitamin D can regulate autophagy at different levels, including
induction, nucleation and elongation to maturation and degradation,
which affects the occurrence and development of diseases (11).
We previously investigated the association between
VDR gene polymorphisms and hypertension. We revealed
polymorphisms in VDRrs11574129, rs2228570 and
rs739837 in 2,409 patients with hypertension and 3,063
controls, and that the rs2228570 polymorphism is
significantly correlated with risk of hypertension (12). However, the mechanism by which
vitamin D signaling regulates blood pressure remains unclear.
Therefore, the present study established a VDR deficiency
animal model using VDR knockout mice to investigate how VDR
regulates blood pressure.
Materials and methods
Animals
VDR-/- mice were derived by
homologous recombination in embryonal stem cells as described
previously (gifted from Dr Marie Demay; Massachusetts General
Hospital, MA, USA) (13).
VDR wild-type (VDR+/+) and knockout
(VDR-/-) mice were identified using western
blotting analysis of tail blood samples (Fig. S1). Animal experiments were
approved by the Institutional Animal Care and Use Committee of
Nanjing Medical University (approval no. IACUC-1910005). All
procedures performed in studies involving animals were in
accordance with the ethical standards of the institution or
practice at which the studies were conducted. For sample
preparation, 100% carbon dioxide was used to euthanize the animals
(14).
Experimental mice were raised in the SPF Animal
Center of Nanjing Medical University, where the room temperature
was maintained at 20-24˚C and ~60% humidity with a 12 h light/dark
schedule. A total of 168-week-old male littermates were randomly
divided into the VDR+/+ and
VDR-/- groups (n=8 mice per group). After
weaning, the mice were fed a regular diet or a ‘rescue diet’
(Harlan Teklad; Envigo) containing 20% lactose, 1.25% phosphorus
and 2% calcium for 8 weeks.
Blood pressure measurements
The ML125 non-invasive blood pressure (NIBP) system
(AD Instruments) was used to measure systolic blood pressure in
conscious animals. A pneumatic pulse sensor cuff was placed on the
tails. After habituation to this setting for 7 days, systolic blood
pressure was recorded. To obtain accurate blood pressure
recordings, the mice were kept in a motionless and undisturbed
state during the measurement. Conditioning was achieved once the
mice were processed gently without forcing restraint. Systolic
blood pressure was recorded consecutively for 3 days in chambers
that were maintained at 31-33˚C. Systolic blood pressure was
recorded separately in 10 min intervals over a total of 10
recordings. The average measurement was calculated for the 10
recordings.
VSMC culture
Primary VSMCs were isolated from aortas of 6- to
8-week-old mice. The mice were euthanized using CO2
(14). After removal of the
adventitia, the aorta was opened to expose the endothelial layer
under a dissection microscope. Tissues from 6 to 8 animals were
pooled and incubated with trypsin (0.25% w/v) at room temperature
for 10 min to remove any remaining adventitia and endothelium.
Tissues were incubated overnight in α Minimum Essential Medium
(αMEM) supplemented with 10% fetal calf serum, 100 U/ml penicillin,
100 µg/ml streptomycin and 0.25 µg/ml amphotericin (complete αMEM)
at room temperature before being digested with 425 U/ml collagenase
type II (Worthington Biochemical Corporation) for 5 h at room
temperature. Isolated VSMCs were expanded in T25 tissue culture
flasks in a humidified atmosphere with 5% CO2 at 37˚C
until reaching confluence. VSMCs were identified by negative
platelet endothelial cell adhesion molecule1 (marker of endothelial
cells) and vimentin (marker of fibroblasts), and positive α-smooth
muscle actin (marker of VSMCs) expression. VSMCs from the 3rd to
5th passages were used in the present study (15).
Reverse transcription-quantitative PCR
(RT-qPCR)
TRIzol® reagent (Invitrogen; Thermo
Fisher Scientific, Inc.) was used to isolate total RNA from
VSMCs according to the manufacturer's instructions.
mRNA levels of SOD, Ang II and Ang II type
1 receptor (AT1R) in the VSMCs were detected using
RT-qPCR (15). Briefly,
first-strand cDNAs were reverse-transcribed from 2 µg of
total RNAs using M-MLV (Moloney Murine Leukemia Virus
Reverse Transcriptase) with oligo(dT); as the primer)
and Rain in 1X M-MLV buffer. Each 1 µl of the cDNA was then
applied as a template for the PCR amplification using the
SYBR® Green PCR reagent kit (Toyobo Life Science) in a
PCR cycler (Applied Biosystems; Thermo Fisher Scientific, Inc.).
GAPDH expression was applied as a loading reference. The target
mRNA was amplified in the following thermocycling conditions:
initial denaturation for 10 min at 95˚C, 40 cycles of denaturation
for 15 sec at 95˚C, annealing for 40 sec at 55˚C, extension for 30
sec at 72˚C and final extension for 7 min at 72˚C. The following
PCR primers were used for corresponding gene detection: Ang-II
(NC_000023.11) forward primer: 5'-CCTCCCGACTAGATGGACAC-3'and
reverse primer: 5'-GAGGGCAGGGGTAAAGAGAG-3'; AT1R (NC_000003.12)
forward primer: 5'-ATGTTTCTTGGTGGCTTGGT-3' and reverse primer:
5'-CCTGAGAGGGTCCGAAGAAA-3'; SOD1 (NC_000021.9) forward primer:
5'-AACCATCCACTTCGAGCAGA-3' and reverse primer:
5'-GGTCTCCAACATGCCTCTCT-3'; GAPDH forward primer:
5'-GAACGGGAAGCTCACTGG-3' and reverse primer:
5'-GCCTGCTTCACCACCTTCT-3'. Relative expression level of the
respective target gene was calculated according to the
2-ΔΔCq method (16).
Western blotting
VSMCs were lysed in RIPA buffer (Sigma-Aldrich;
Merck KGaA) containing proteinase inhibitors (Sigma-Aldrich). The
protein concentration of the lysate was determined using a BCA kit
(Merck KGaA). Aliquots (20 µg) of the extracted protein sample were
boiled for 5 min, loaded on an 10% SDS-PAGE gel, separated by
electrophoresis and then transferred onto a nitrocellulose
membrane. The membrane was blocked with 5% milk at room temperature
in phosphate-buffered saline/0.05% Tween 20 (PBST) for 3 h and then
incubated with a monoclonal antibody against renin (1:1,000; cat.
no. 70R-1584; Fitzgerald) overnight at 4˚C. After three washes with
PBST, the membrane was incubated with horseradish
peroxidase-conjugated secondary antibodies (1:1,000; cat. no.
A0208; Beyotime Institute of Biotechnology) at room temperature for
1 h. Finally, the probed bands were visualized using an Enhanced
Chemiluminescence reagent (PerkinElmer) and analyzed using ImageJ
(version 1; National Institutes of Health) (15). Other used antibodies were as
follows: anti-LC3-A (cat. no. AF5225; 1:1,000), anti-Beclin1 (cat.
no. AF5123; 1:1,000), anti-AGT7 (cat. no. AA820; 1:1,000), and
anti-GAPDH (cat. no. AF0006; 1:1,000) were purchased from Beyotime
Institute of Biotechnology.Anti-ATR1 (cat. co. PB0492; 1:1,000),
anti-P62 (cat. no. BA2849; 1:1,000), and anti-PRDX4 (cat. co.
PB9383; 1:1,000) were purchased from BOSTER Institute of
Biotechnology. GAPDH was used as a loading control.
Transmission electron microscopy
(TEM)
For cellular TEM observation, VSMCs were cultured
for 120 min and then fixed with 2.5% glutaraldehyde and post-fixed
with 3% osmium tetroxide for 2 h at room temperature. The specimen
was dehydrated in a graded series of ethanol, embedded with
EPon812, and stained by uranium acetate and aluminum citratein.
Epon resin and then observed with a Hitachi-600 TEM (Hitachi, Ltd.)
to evaluate the formation of autophagosomes in the cells (17).
Statistical analysis
All data were statistically analyzed using SPSS
software (version 13.0; SPSS, Inc.). In the present study, all data
are presented as the mean ± standard deviation. Average data
between the groups was compared using the unpaired Student's
t-test. The Levene test was applied for distribution analysis.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Systolic blood pressure is elevated in
VDR-/- mice
Systolic blood pressure was measured using the NIBP
system in all mice. The systolic blood pressure of the
VDR-/- mice was significantly higher compared
with the VDR+/+ littermate control mice (Fig. 1).
The RAS is upregulated in
VDR-/- mice
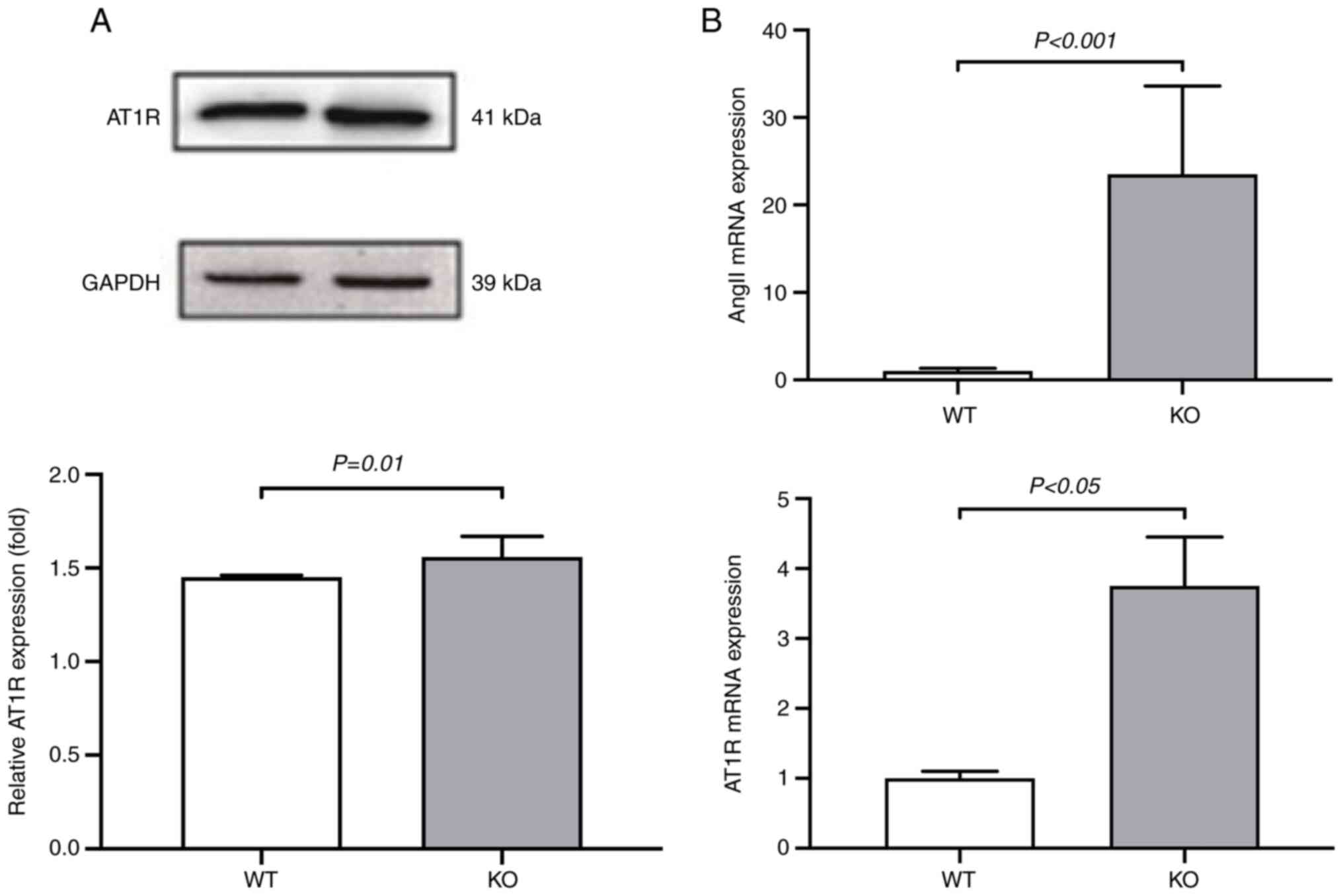
To understand how VDR deficiency affects
hypertension and the RAS, the expression levels of RAS factors Ang
II and AT1R were measured using western blotting and RT-qPCR assays
in VSMCs isolated from both VDR-/- and
VDR+/+ mice. Protein expression of AT1R was
significantly increased in the VDR-/- VSMCs
compared with the VDR+/+ VSMCs (Fig. 2A). The mRNA levels of Ang II
and AT1R were significantly upregulated in the
VDR-/- VSMCs compared with the
VDR+/+ VSMCs (Fig.
2B). These results suggested that deletion of VDR upregulated
the RAS in mice.
Oxidative stress is elevated in
VDR-/- mice
The association between hypertension in the
VDR-/- mice and oxidative stress levels were
determined in the VSMCs. mRNA levels of SOD were measured
using RT-qPCR and protein expression of Prdx4 was measured using
western blotting analysis in the VSMCs isolated from both
VDR-/- and VDR+/+ mice. The
results demonstrated that the mRNA levels of SOD were
significantly downregulated in the VDR-/- VSMCs
compared with the VDR+/+ VSMCs (Fig. 3A). The protein expression of Prdx4
was significantly decreased in the VDR-/- VSMCs
compared with the VDR+/+ VSMCs (Fig. 3B). These data indicated that
VDR deficiency upregulated oxidative stress in mice.
Protein expression levels of
autophagy-related factors are upregulated in VSMCs of
VDR-/- mice
The expression levels of autophagy-related factors,
including autophagy-related protein 7 (ATG7), Beclin1,
microtubule-associated proteins 1A/1B light chain 3A(LC3A) and
nucleoporin p62 (p62), were measured in the VSMCs of
VDR-/- and VDR+/+ mice using
western blotting analysis. ATG7, Beclin1 and LC3Awere significantly
upregulated, while p62 was significantly downregulated in the
VDR-/- VSMCs compared with the
VRD+/+ VSMCs (Fig.
4).
 | Figure 4Protein expression of ATG7, Beclin1,
LC3A and p62 in VSMCs. Protein expression of ATG7, Beclin1, LC3A
and p62 in VSMCs isolated from VDRKO and VDRWT mice
was detected using western blotting analysis. (A) Representative
images of the corresponding western blot images. The average
intensities of (B) ATG7, (C) p62, (D) Beclin1 and (E) LC3A are
summarized in the corresponding bar graphs from three independent
assays. WT levels were set at 1.0 for data normalization. KO,
knockout/VDR-/-; WT,
wild-type/VDR+/+; VSMCs, vascular smooth muscle
cells; ATG7, autophagy-related protein 7; p62, nucleoporin
p62; LC3A, microtubule-associated proteins 1A/1B light chain
3A; VDR, vitamin D receptor. |
TEM reveals increased autophagosomes
in VSMCs of VDR-/- mice
Next, the VSMC ultrastructure in the
VDR-/- and VDR+/+ mice was
analyzed using TEM. An increased number of autophagy bodies were
observed in the VDR-/- VSMCs compared with the
VDR+/+ VSMCs (Fig.
5). These results suggested that VDR deficiency could
activate autophagy in VSMCs.
Discussion
Oxidative stress can injure blood vessels and serve
as a pathogenic factor in hypertension. Numerous studies have
demonstrated that there is an imbalance between the anti-oxidative
defense system and the production of oxygen free radicals, causing
a high level of oxidative stress in patients with hypertension
(18-20).
Dysfunction of vascular endothelial cells caused by oxidative
stress is considered to be the main cause of hypertension (21). Oxidative stress is closely
associated with endothelial cell inflammation, hypertrophy,
apoptosis, migration, fibrosis and vascular remodeling in
hypertension (19,22).
Since the VDR is widely distributed in vascular
endothelial cells, VSMCs and cardiomyocytes, the role of VDR in
hypertension has received extensive attention. In a previous
observational study, activation of the VDR is associated with lower
cardiovascular risk and improved survival (23). VDR deficiency can elevate
intracellular oxidative stress (23), and VDR agonists have been
demonstrated to synergistically alleviate diabetic atherosclerosis
by inhibiting oxidative stress (24). Consistent with previous findings,
the present study revealed that SOD mRNA levels and Prdx4
protein expression were significantly downregulated in
VDR-/- mice compared with the VDR+/+
mice. These data suggested that VDR deficiency could increase
oxidative stress.
Oxidative stress reflects an imbalance between the
reactive oxygen species and a biological ability to detoxify or
repair the resulting damage. SOD is an enzyme that downregulates
O2-byscavenging potentially damage-free radical
moieties. It acts as a major anti-oxidative enzyme in almost all
organisms. Therefore, SOD level reflects the anti-oxidative
capacity. The higher the level of SOD, the higher capacity of
anti-oxidation, which results the positive balance of
anti-oxidation and pro-oxidation. Oppositely, the lower level of
the SOD, the lower capacity of anti-oxidation, which causes the
negative balance of anti-oxidation and pro-oxidation or oxidative
damage (23). The results of the
present study revealed that SOD level was decreased in the
VDR-/- mice, which indirectly reflects the
upregulated oxidative stress.
Indeed, this pattern is consistent with a number of
previous findings. For example, in primary angle closure glaucoma,
oxidative stress is increased accompanied with a decrease of SOD
level (25). Under normal
circumstances, production and clearance of reactive oxygen species
(ROS) are in equilibrium. However, once ROS production exceeds
clearance, a large number of oxygen free radicals will be generated
in the body. In patients with hypertension, increased production of
ROS results in decreased levels of SOD, destruction of unsaturated
fatty acids and increased lipid peroxidation, causing increased
production of malondialdehyde.
Vitamin D signaling plays an important role in the
inhibition of renin secretion and synthesis. Disruption of VDR
signaling transduction leads to RAS activation, cardiac hypertrophy
and hypertension (26). It has
been demonstrated that VDR-/- mice, a model of
vitamin D signal disruption, develop hypertension (26). Vitamin D inhibits the
renin-angiotensin-aldosterone system by blocking renin gene
expression (3). Plasma renin and
Ang II levels are negatively correlated with
1,25(OH)2D3 (27). Knockout of VDR and cytochrome P450
27B1 in mice results in elevated serum renin and RAS activity and
increased blood pressure (5).
Xiang et al (28) reported
an increase in renin and Ang II mRNA levels in the hearts of
1α(OH)ase and VDR knockout mice. The
present study demonstrated that AT1R and Ang II levels increased
significantly in VDR-/- VSMCs, consistent with
the previous findings. Vitamin D can inhibit renin gene expression
by activating the cAMP response elements at the promoter region of
the renin gene (4). Overexpression
of VDR can inhibit renin production in renal par acyclic cells
(29). Based on the result of the
present study and the literature, we hypothesize that VDR
deficiency induces overexpression of renin, thus activating the RAS
in mouse VSMCs. However, further research is needed to confirm this
hypothesis.
Autophagy plays an important role in human health.
Numerous disorders are associated with autophagy imbalances, such
as hypertension and cardiac disease (30). Vitamin D has been reported to
regulate autophagy through multiple pathways, including gene
induction, nucleation and elongation of protein maturation and
degradation (31). However, the
mechanism by which the VDR regulates autophagy has not been fully
determined. An improved understanding of this mechanism could be
useful for clinical diagnosis and treatment of relative
diseases.
To the best of our knowledge, thus far, studies on
vitamin D-mediated regulation of autophagy have mainly focus on the
phosphatidylinositol 3 kinase/Beclin-1 pathway, Ca2+
levels, toll-like receptor signaling pathway, antimicrobial
peptides and lysosomes, autophagy related gene expression and
inflammatory factors (31). Ang
II, a vasoactive peptide, plays a notable role in numerous vascular
disorders. An imbalance in vascular autophagy, excessive VSMC
proliferation and vascular remodeling can lead to increased
vascular resistance and lumen stenosis, resulting in increased
blood pressure (32,33). Hypertensive rats demonstrated
endothelial dysfunction in aortic and mesenteric arteries, with
decreased phosphorylated (p)-Akt, p-mTOR and autophagy marker
protein p62, and increased LC3 II/I levels (34). Ang II and glomerular podocyte
autophagic activity increased significantly in hypertensive rat
kidneys; therefore, high blood pressure caused by kidney injury may
be associated with Ang II-induced glomerular podocyte autophagy.
Excessive autophagy causes endothelial dysfunction in rats, but it
has been revealed that endothelial function can be improved and
blood pressure can be reduced through regulating autophagy
(35). Ang II induces autophagy
through the IAT1R/Rhoda/Rho kinase pathway, causing hypertrophy of
VSMCs (36). The interaction
between autophagy, oxidative stress and the RAS plays a notable
role in vascular remodeling and vascular damage caused by
hypertension (37,38).
The objective of the present study was to explore
the possible mechanism of VDR deficiency on the RAS and cellular
autophagy in a mouse model of vitamin D deficiency. The present
study data demonstrated that VDR deficiency increased oxidative
stress via downregulating SOD levels and Prdx4 expression,
and activating autophagy via upregulation of ATG7, Beclin1 and
LC3Aexpression levels in VSMCs. We hypothesize that the increased
autophagy level induced by VDR deficiency may be associated
with activation of the RAS and signaling pathways downstream of
oxidative stress.
In conclusion, the present study suggested that
VDR deficiency increased blood pressure by elevating
oxidative stress factors and RAS activity, in addition to causing
excessive autophagy of VSMCs. The present study offered novel
insight into the mechanism by which VDR regulates blood pressure
and provided theoretical evidence to guide clinicians on
administering 1,25(OH)2D3 for the prevention
and treatment of hypertension.
Supplementary Material
VDR KO mouse verification.
Blood samples from VDRKO and WT mice were obtained from mice
tails. VDR protein expression was detected using western blotting
analysis. GAPDH protein expression was used as a control to
normalize VDR expression. VDR, vitamin D receptor; KO,
knockout/VDR-/-; WT,
wild-type/VDR+/+; NC, negative control.
Acknowledgements
The authors thank Dr Marie Demay (Massachusetts
General Hospital, MA, USA) for supplying the
VDR-/- mice for this study.
Funding
Funding: This study was supported by Young Scholars Fostering
fund of the First Affiliated Hospital of Nanjing Medical University
(PY2021015); Jiangsu Province ‘Six Talent Peaks’ High-Level Talent
Project (grant no. WSN-024); and Jiangsu Research Center for
Primary Health Development and General Practice Education (grant
no. 2019B03).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YZ performed the research conception and design. XT,
JL, YZ and XY performed the experiments. JJ and ZT analyzed and
checked the data, and drafted the manuscript. JJ, ZT and XY
prepared figures. JJ and YZ edited and revised manuscript. YZ was
primarily responsible for final content. JJ and YY confirm the
authenticity of all the raw data. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
Animal experiments were approved by the
Institutional Animal Care and Use Committee of Nanjing Medical
University (approval no. IACUC-1910005). All procedures performed
in studies involving animals were in accordance with the ethical
standards of the institution or practice at which the studies were
conducted.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Latic N and Erben RG: Vitamin D and
cardiovascular disease, with emphasis on hypertension,
atherosclerosis, and heart failure. Int J Mol Sci.
21(6483)2020.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Tamez H, Kalim S and Thadhani RI: Does
vitamin D modulate blood pressure? Curr Opin Nephrol Hypertens.
22:204–209. 2013.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Yuan W, Pan W, Kong J, Zheng W, Szeto FL,
Wong KE, Cohen R, Klopot A, Zhang Z and Li YC:
1,25-dihydroxyvitamin D3 suppresses renin gene transcription by
blocking the activity of the cyclic AMP response element in the
renin gene promoter. J Biol Chem. 282:29821–29830. 2007.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Zhou C, Lu F, Cao K, Xu D, Goltzman D and
Miao D: Calcium-independent and 1,25(OH)2D3-dependent regulation of
the renin-angiotensin system in 1alpha-hydroxylase knockout mice.
Kidney Int. 74:170–179. 2008.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Andersen LB, Przybyl L, Haase N, von
Versen-Höynck F, Qadri F, Jørgensen JS, Sorensen GL, Fruekilde P,
Poglitsch M, Szijarto I, et al: Vitamin D depletion aggravates
hypertension and target-organ damage. J Am Heart Assoc.
4(e001417)2015.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Ji S, Doumit ME and Hill RA: Regulation of
adipogenesis and key adipogenic gene expression by 1,
25-dihydroxyvitamin D in 3T3-L1 cells. PLoS One.
10(e0126142)2015.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Yin Y, Yu Z, Xia M, Luo X, Lu X and Ling
W: Vitamin D attenuates high fat diet-induced hepatic steatosis in
rats by modulating lipid metabolism. Eur J Clin Invest.
42:1189–1196. 2012.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Asano L, Watanabe M, Ryoden Y, Usuda K,
Yamaguchi T, Khambu B, Takashima M, Sato SI, Sakai J, Nagasawa K
and Uesugi M: Vitamin D metabolite, 25-hydroxyvitamin D, regulates
lipid metabolism by inducing degradation of SREBP/SCAP. Cell Chem
Biol. 24:207–217. 2017.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Wimalawansa SJ: Vitamin D deficiency:
Effects on oxidative stress, epigenetics, gene regulation, and
aging. Biology (Basel). 8(30)2019.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Fujii J, Ikeda Y, Kurahashi T and Homma T:
Physiological and pathological views of peroxiredoxin 4. Free Radic
Biol Med. 83:373–379. 2015.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Liu L, Li J, Deng C and Chen D: Advances
in the mechanism of vitamin D affecting autophagy. Zhonghua Wei
Zhong Bing Ji Jiu Yi Xue. 30:1103–1106. 2018.PubMed/NCBI View Article : Google Scholar : (In Chinese).
|
|
12
|
Jia J, Shen C, Mao L, Yang K, Men C and
Zhan Y: Vitamin D receptor genetic polymorphism is significantly
associated with decreased risk of hypertension in a Chinese Han
population. J Clin Hypertens (Greenwich). 16:634–639.
2014.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Li YC, Pirro AE, Amling M, Delling G,
Baron R, Bronson R and Demay MB: Targeted ablation of the vitamin D
receptor: An animal model of vitamin D-dependent rickets type II
with alopecia. Proc Natl Acad Sci USA. 94:9831–9835.
1997.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Boivin GP, Hickman DL, Creamer-Hente MA,
Pritchett-Corning KR and Bratcher NA: Review of CO2 as a
euthanasia agent for laboratory rats and mice. J Am Assoc Lab Anim
Sci. 56:491–499. 2017.PubMed/NCBI
|
|
15
|
Patel JJ, Bourne LE, Millán JL, Arnett TR,
MacRae VE, Wheeler-Jones CPD and Orriss IR: Inhibition of vascular
smooth muscle cell calcification by ATP analogues. Purinergic
Signal. 15:315–326. 2019.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Zhang J, Yuan L, Wang S, Liu J, Bi H, Chen
G, Li J and Chen L: Germacrone protects against oxygen-glucose
deprivation/reperfusion injury by inhibiting autophagy processes in
PC12 cells. BMC Complement Med Ther. 20(77)2020.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Allison SJ: Hypertension: Oxidative stress
and immune activation in hypertension. Nat Rev Nephrol.
12(4)2016.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Sinha N and Dabla PK: Oxidative stress and
antioxidants in hypertension-a current review. Curr Hypertens Rev.
11:132–142. 2015.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Guzik TJ and Touyz RM: Oxidative stress,
inflammation, and vascular aging in hypertension. Hypertension.
70:660–667. 2017.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Wang D, Strandgaard S, Iversen J and
Wilcox CS: Asymmetric dimethylarginine, oxidative stress, and
vascular nitric oxide synthase in essential hypertension. Am J
Physiol Regul Integr Comp Physiol. 296:R195–R200. 2009.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Irani K: Oxidant signaling in vascular
cell growth, death, and survival: A review of the roles of reactive
oxygen species in smooth muscle and endothelial cell mitogenic and
apoptotic signaling. Circ Res. 87:179–183. 2000.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Argacha JF, Egrise D, Pochet S, Fontaine
D, Lefort A, Libert F, Goldman S, van de Borne P, Berkenboom G and
Moreno-Reyes R: Vitamin D deficiency-induced hypertension is
associated with vascular oxidative stress and altered heart gene
expression. J Cardiovasc Pharmacol. 58:65–71. 2011.PubMed/NCBI View Article : Google Scholar
|
|
24
|
He L, He T, Farrar S, Ji L, Liu T and Ma
X: Antioxidants maintain cellular redox homeostasis by elimination
of reactive oxygen species. Cell Physiol Biochem. 44:532–553.
2017.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Li S, Shao M, Li Y, Li X, Wan Y, Sun X and
Cao W: Relationship between oxidative stress biomarkers and visual
field progression in patients with primary angle closure glaucoma.
Oxid Med Cell Longev. 2020(2701539)2020.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Kong J and Li YC: Effect of ANG II type I
receptor antagonist and ACE inhibitor on vitamin D receptor-null
mice. Am J Physiol Regul Integr Comp Physiol. 285:R255–R261.
2003.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Tomaschitz A, Pilz S, Ritz E, Grammer T,
Drechsler C, Boehm BO and März W: Independent association between
1,25-dihydroxyvitamin D, 25-hydroxyvitamin D and the
renin-angiotensin system: The ludwigshafen risk and cardiovascular
health (LURIC) study. Clin Chim Acta. 411:1354–1360.
2010.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Xiang W, Kong J, Chen S, Cao LP, Qiao G,
Zheng W, Liu W, Li X, Gardner DG and Li YC: Cardiac hypertrophy in
vitamin D receptor knockout mice: Role of the systemic and cardiac
renin-angiotensin systems. Am J Physiol Endocrinol Metab.
288:E125–E132. 2005.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Kong J, Qiao G, Zhang Z, Liu SQ and Li YC:
Targeted vitamin D receptor expression in juxtaglomerular cells
suppresses renin expression independent of parathyroid hormone and
calcium. Kidney Int. 74:1577–1581. 2008.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Galluzzi L and Green DR:
Autophagy-independent functions of the autophagy machinery. Cell.
177:1682–1699. 2019.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Tai S, Hu XQ, Peng DQ, Zhou SH and Zheng
XL: The roles of autophagy in vascular smooth muscle cells. Int J
Cardiol. 211:1–6. 2016.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Salabei JK and Hill BG: Implications of
autophagy for vascular smooth muscle cell function and plasticity.
Free Radic Biol Med. 65:693–703. 2013.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Justin Rucker A and Crowley SD: The role
of macrophages in hypertension and its complications. Pflugers
Arch. 469:419–430. 2017.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Dong Q, Xing W, Fu F, Liu Z, Wang J, Liang
X, Zhou X, Yang Q, Zhang W, Gao F, et al: Tetrahydroxystilbene
glucoside inhibits excessive autophagy and improves microvascular
endothelial dysfunction in prehypertensive spontaneously
hypertensive rats. Am J Chin Med. 44:1393–1412. 2016.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Dong Q, Xing W, Su F, Liang X, Tian F, Gao
F, Wang S and Zhang H: Tetrahydroxystilbene glycoside improves
microvascular endothelial dysfunction and ameliorates
obesity-associated hypertension in obese ZDF rats via inhibition of
endothelial autophagy. Cell Physiol Biochem. 43:293–307.
2017.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Mondaca-Ruff D, Riquelme JA, Quiroga C,
Norambuena-Soto I, Sanhueza-Olivares F, Villar-Fincheira P,
Hernández-Díaz T, Cancino-Arenas N, San Martin A, García L, et al:
Angiotensin II-regulated autophagy is required for vascular smooth
muscle cell hypertrophy. Front Pharmacol. 9(1553)2018.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Wang ZV, Rothermel BA and Hill JA:
Autophagy in hypertensive heart disease. J Biol Chem.
285:8509–8514. 2010.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Zhou L, Ma B and Han X: The role of
autophagy in angiotensin II-induced pathological cardiac
hypertrophy. J Mol Endocrinol. 57:R143–R152. 2016.PubMed/NCBI View Article : Google Scholar
|