Introduction
The development of the lens in the eye begins with
the PAX6-expressing epidermal ectoderm making contact with the
optic vesicle. Subsequently, SOX2 is expressed in the ectoderm
region in contact with the optic vesicle, and the coordinated
action of PAX6 as a partner factor for SOX2 causes cells in contact
with the optic vesicle to thicken and form lens placodes, which are
depressed inward by the formation of the optic cup (1). The lens placode is eventually
separates from the epidermal ectoderm to form a spherical lens
vesicle with an array of surrounding cells. Moreover, the cells
that are positioned on the side of the lens follicle extend toward
the interior of the lens placode and fill the interior (2,3).
The human lens is 9-10 mm in diameter and is
surrounded by a lens capsule, which is rich in type IV collagen. On
the corneal side, the lens epithelium is composed of a single layer
of lens epithelial cells (LECs). Around the equatorial region of
the lens, LECs separate from the capsule and begin to elongate
toward the anterior and posterior poles of the lens, where the LECs
differentiate into lens fiber cells (LFCs). Eventually, the
organelles inside the LFCs disappear (4) and are replaced by lens fibers filled
with crystallin proteins. At the center of the lens lies the lens
nucleus (fetal nucleus), which is formed during development and is
surrounded by lens fibers (5).
After birth, LECs continue to proliferate slowly, and the few
remaining lens-tissue stem cells expressing p75NTR are suspected to
be involved in proliferation (6).
However, the proliferation of these cells slows with age (7).
The lens is composed of approximately 90%
crystallin, a water-soluble protein. Crystallin in vertebrates is
mainly classified into α-, β-, and γ-crystallin. α-crystallin has 2
subunits of αA- and αB-, β-crystallin has 7 subunits of βA1-βA4 and
βB1-βB3, and γ-crystallin has 5 subunits of γA-γD and γS (8). In the lens, α-crystallin is a 40-mer,
β-crystallin is a 2-6-mer, and γ-crystallin is a monomer, which
play an important role in interacting with each other to maintain
the transparency of the lens (9).
α-crystallin is a major component of the lens, accounts for
approximately 30% of the water-soluble protein in the lens, and
functions as a molecular chaperone that suppresses aggregation of
other crystallin species (10). In
humans, αB-crystallin is expressed in tissues other than the eye,
whereas αA-crystallin is expressed only in the lens (11). βB2-crystallin is the main component
of β-crystallin. It has been reported that in βB2-crystallin
obtained from the lens of senile cataract, the Asp residue at the
C-terminal site undergoes significant site-specific isomerization,
which occurs at the site of interaction with βB2-crystallin itself
and other βB-crystallins, and it may contribute to the formation of
senile cataract by affecting the crystallin subunit-subunit
interaction and inducing abnormal crystallin aggregation (12).
After the lens of a newt is removed, the pigmented
epithelial cells (PECs) located on the dorsal side of the iris
first start to dedifferentiate, a process during which their
pigment is degranulated, and then differentiate into LECs to
regenerate a new lens (13,14).
By contrast, regeneration of the lens in mammals does not occur
once the lens capsule is removed (15). Intriguingly, if only the contents
of the lens are removed and the lens capsule and LECs are
preserved, the lens reproduces the same process as that occurring
during embryonic development and forms a regenerated lens (16-18).
However, the regenerated LECs exhibit aberrant, morphological
changes, including irregular cell arrangement, mitochondrial
degeneration, and vacuoles in the cytoplasm (19).
Induced pluripotent stem (iPS) cells, first
described by Takahashi et al (20), are pluripotent cells that can
differentiate into diverse cell types. Notably, iPS cells have also
been reported to differentiate into LECs (21-24),
but no study thus far has reported their formation of
three-dimensional cell aggregates. Here, we investigated the
formation of three-dimensional cellular aggregates expressing
lens-specific proteins using human iris-derived tissue cells and
iPS cells.
Materials and methods
Preparation of human iris tissue
specimens
The tissues examined in the study were collected
from patients with glaucoma during treatment with partial iris
resection, and pieces of the collected human iris tissue were fixed
in SUPER FIX™ rapid fixative solution (cat. no. KY-500;
Kurabo Industries Ltd.) (6).
Subsequently, paraffin sections were prepared from the fixed
tissues by following standard procedures, and the sections were
stained with hematoxylin and eosin (H&E). Iris samples were
collected from patients who underwent partial iris resection as a
treatment for glaucoma at the Department of Ophthalmology, Fujita
Health University Hospital, between April 2015 and March 2017, and
who consented to the study. The mean age of the patients was
58.9±5.4 years, 4 males and 7 females. Patients with ocular
diseases other than glaucoma were excluded from the study. This
study was performed with the approval (approval no. 05-065) of the
Ethics Review Committee of Fujita Health University. The experiment
was carried out with the approval (approval no. DP16055) of the
Recombinant DNA Experiment Committee of Fujita Health University.
All study participants provided written informed consent for their
tissue to be used, and the study complied with the tenets of the
Declaration of Helsinki for research involving human tissues.
Isolation and culture of cells from
human iris tissue
Human iris tissue was processed as described
(25,26). Briefly, iris tissue was treated
with 0.2% collagenase (cat. no. C9722-50MG; Merck KGaA) and washed
twice with phosphate-buffered saline (PBS; cat. no. D8662-500ML;
Merck KGaA), and the isolated human iris tissue-derived cells
(H-iris cells) were cultured in iris culture medium (iris medium):
Advanced Dulbecco's modified Eagle's medium/Ham's F12 (Advanced
DMEM/F12; cat. no. 12634010; Thermo Fisher Scientific Inc.)
supplemented with 5% (w/v) mixed serum [heat-inactivated human
serum (cat. no. H3667-20ML; Merck KGaA), KnockOut™ Serum
Replacement (KSR; cat. no. 10828010; Thermo Fisher Scientific), and
Artificial Serum, Xeno-free (cat. no. A2G10P2CC; Cell Science &
Technology Institute, Inc.) in a 5:3:2 ratio], 10 ng/ml basic
fibroblastic growth factor (b-FGF; cat. no. F0291; Merck KGaA), 10
ng/ml epidermal growth factor (cat. no. E9644; Merck KGaA), 1%
(w/v) GlutaMAX™ (cat. no. 35050061; Thermo Fisher
Scientific), 0.1% (w/v) CultureSure® Y-27632 solution
(used only when starting the culture; cat. no. 039-24591; FUJIFILM
Wako Pure Chemical Corporation), and 1% (w/v)
penicillin/streptomycin (cat. no. P4458-100ML; Merck KGaA). The
cells were plated in culture dishes coated with type I collagen
(cat. no. TMTCC-050; Toyobo Co., Ltd.) and incubated at 37˚C in a
5% CO2 humidified incubator, and the cultured cells were
examined using an inverted fluorescence microscope equipped with a
digital camera system (Power IX-71 and DP-71; Olympus
Corporation).
Preparation of human iris-derived iPS
cells
Human iris-derived iPS (H-iris iPS) cells were
prepared through cell reprogramming using H-iris cells, as
described previously (26).
Briefly, H-iris cells were reprogrammed by employing a
micro-electroporation method performed using an Epi5™
Episomal iPSC Reprogramming Kit (cat. no. A15960; Thermo Fisher
Scientific) (27). After cloning,
StemFit® (cat. no. RCAK02N; ReproCELL Inc.) mixed with
0.1% (w/v) CultureSure® Y-27632 solution (only at the
beginning of culture) was used as the iPS cell-culture medium, and
iMatrix-511 (Laminin-5; cat. no. 892011; Takara Bio Inc.) was used
as the coating agent (STEP-0 culture condition; Fig. 1). During passaging, the H-iris iPS
cells were detached using a mixture of Accutase (cat. no.
AT104-100ML; M&S TechnoSystems, Inc.) and TrypLE Select Enzyme
(cat. no. 12563011; Thermo Fisher Scientific). The H-iris cells and
H-iris iPS cells were confirmed to be negative for mycoplasma
infection using a mycoplasma detection kit (EZ-PCR™
Mycoplasma Test Kit, cat. no. 20-700-20; Biological Industries USA
Inc.) according to the manufacturer's instructions. The experiment
was conducted with the approval (no. DP16055) of the Recombinant
DNA Experiment Committee of Fujita Medical University.
Differentiation of H-iris cells and
H-iris iPS cells into LECs
The composition of the used LEC medium (with the
STEP-4 culture condition being the same; Fig. 1), which induces H-iris cells to
differentiate into LECs, was as follows (28): 10% (w/v) fetal bovine serum (cat.
no. 04-111-1A; Biological Industries Israel Beit-Haemek), 10 ng/ml
b-FGF, 1% (w/v) minimum essential medium non-essential amino acids
(cat. no. 11140050; Thermo Fisher Scientific), 0.5% (w/v)
GlutaMAX™, DMEM high-glucose medium (cat. no. 11965092;
Thermo Fisher Scientific), and 1% (w/v) penicillin/streptomycin
solution.
The common differentiation culture conditions were
based on reports describing the differentiation of human embryonic
stem (ES) cells into lens progenitor cells and lentoid bodies
(29); H-iris iPS cells were
cultured in Lens differentiation base medium: Advanced DMEM/F12
supplemented with 0.05% (w/v) bovine serum albumin (cat. no.
012-23881; FUJIFILM Wako), 1% (w/v) non-essential amino acids
(Thermo Fisher Scientific), 0.5% (w/v) GlutaMAX™, 0.5%
(w/v) N-2 MAX Media Supplement (cat. no. 17502048; Thermo Fisher
Scientific), 1% (w/v) B-27 supplement (cat. no. 17504044; Thermo
Fisher Scientific), 100 ng/ml b-FGF, and 0.1% (w/v)
CultureSure® Y-27632 solution, with 100 ng/ml Noggin
(cat. no. 6057-NG; R&D Systems, Inc.) added for 6 days
(STEP-1). On the 6th day, the medium was replaced with lens
differentiation base medium containing 20 ng/ml bone morphogenetic
protein-4/7 (cat. no. 3727-BP; R&D Systems) and 100 ng/ml b-FGF
(STEP-2), and on the 18th day, this medium was replaced with lens
differentiation base medium containing 20 ng/ml Wnt-3a (cat. no.
5036-WN; R&D Systems) and 100 ng/ml b-FGF (STEP-3).
Distinct culture environments were used in our
experiments, Experiments 1-5 (Ex.1-Ex.5), as described below
(summarized in Fig. 1): Ex.1:
1x105 H-iris iPS cells were seeded into 3.5 cm adhesive
cell-culture dishes coated with iMatrix-511 and cultured in iPS
culture medium at 37˚C in a 5% CO2 humidified incubator,
and starting from the next day, the cells were maintained as
adherent cultures sequentially under STEP-1, STEP-2, and STEP-3
culture conditions. Ex.2: 1x105 H-iris iPS cells were
seeded into 3.5 cm adhesive cell-culture dishes coated with
iMatrix-511 and cultured in iPS culture medium at 37˚C in a 5%
CO2 humidified incubator, and from the next day, the
cells were maintained as adherent cultures sequentially under
STEP-1, STEP-2, and STEP-3 culture conditions. Under STEP-3
conditions applied for the last 10 days, the cells were detached
using a mixture of Accutase and TrypLE Select Enzyme and cultured
using the inclined rotational-suspension method in a centrifuge
tube. Ex.3: 1x106 H-iris iPS cells were cultured
sequentially under the culture conditions of STEP-1 using the
static-suspension method, STEP-2 using the inclined
rotational-suspension method, and STEP-3 using the horizontal
rotational-suspension method in a centrifuge tube. Ex.4:
1x106 H-iris iPS cells were cultured in a centrifuge
tube under STEP-1, STEP-2, and STEP-3 culture conditions
sequentially using the static-suspension method and then using the
inclined rotational-suspension method under STEP-4 conditions for 2
weeks. Ex.5 is described in the next subsection. The medium was
changed every 2 days in Ex.1-Ex.5. For inclined rotation in the
experiments, an NRC20D rotary mixer (Nissinrika Co., Ltd., Tokyo,
Japan) was used at 3 rpm and a tilt angle of 45˚, and for
horizontal rotation, an RT-50 rotator (TAITEC Corporation) was used
at 35 rpm.
Differentiation of multiple ocular
cells
As reported by Hayashi et al (30), H-iris iPS cells were cultured using
the self-formed ectodermal autonomous multi-zone (SEAM) method for
ocular cells (Ex.5). Briefly, H-iris iPS cells were seeded into
iMatrix-511-coated cell-culture dishes at 350 cells/cm2
using the StemFit® medium, and at 4 weeks after seeding,
the culture medium (SEAM medium) was changed to the following
differentiation medium: DMEM (cat. no. D5796-500ML; Merck KGaA)
supplemented with 10% (w/v) KSR, 1% (w/v) sodium pyruvate (cat. no.
11360070; Thermo Fisher Scientific), 1% (w/v) non-essential amino
acids, 1% (w/v) GlutaMAX™, 1% (w/v) monothioglycerol
(cat. no. 195-15791; FUJIFILM Wako), and 1% (w/v)
penicillin/streptomycin; the cells were cultured at 37˚C in a 5%
CO2 humidified incubator. The cells present in the area
where LECs were observed using the SEAM method were harvested and
cultured using the rotational-suspension method. After 4 weeks, the
cell aggregates that formed between the 2nd and 3rd zones of the
SEAM were picked using a pipette, cloned, and cultured in LEC
medium, and then 1x106 proliferated cells were cultured
under the same conditions as in Ex.3 and differentiated into
LECs.
Reverse transcription and quantitative
PCR (qPCR)
Total cellular RNA was extracted using a
TaqMan® Gene Expression Cells-to-CT™ Kit
(cat. no. A25603; Thermo Fisher Scientific), and RNA concentrations
were measured using a spectrophotometer (NanoVue™; GE
Healthcare) (31). Total RNA was
reverse-transcribed using a GeneAmp® PCR System 9700
Thermal Cycler (Thermo Fisher Scientific) to synthesize cDNA, and,
subsequently, qPCR was performed using an ABI PRISM®
7900 HT Sequence Detection System (Thermo Fisher Scientific) and
the following primers and probes (TaqMan® Gene
Expression assays; cat. no. 4331182; Thermo Fisher Scientific):
p75NTR (assay ID. Hs00609976_m1), SOX2 (assay ID.
Hs00415716_m1), PAX6 (assay ID. Hs01088114_m1), type IV
collagen (assay ID. Hs00266237_m1), and the crystalline
lens-marker gene αA-crystallin (assay ID. Hs00166138_m1);
glyceraldehyde-3-phosphate dehydrogenase (GAPDH; assay ID.
Hs99999905_m1) was used as an internal positive control. The
thermocycling conditions were as follows: reverse transcription: 60
min at 37˚C and 5 min at 95˚C. qPCR: 2 min at 50˚C, 10 min at 95˚C,
15 sec at 95˚C and 1 min at 60˚C, for 50 cycles. Relative
expression was analyzed by the delta-delta Ct method using Ct
values obtained from qPCR amplification.
Sectioned specimens of cell
aggregates
Cell aggregates were collected from suspension
cultures and treated using the cell-block method (32) to prepare paraffin-section specimens
using a fixative solution as described previously.
Immunofluorescence staining
Immunofluorescence staining was performed as
previously described (26,33). Briefly, fixed cells were
permeabilized with 0.5% Triton X-100 (cat. no. 04605-250; FUJIFILM
Wako), blocked with a serum-free ready-to-use blocking reagent
(cat. no. X090930-2; Agilent Technologies, Inc.) for 5 min at room
temperature, and stained with one of the following primary
antibodies (incubated for 1 h at 37˚C): anti-human αA-crystallin
rabbit polyclonal antibody (1:100; cat. no. ab5595; Abcam plc.),
anti-human PAX6 mouse monoclonal antibody (1:100; cat. no.
14-9914-80; Thermo Fisher Scientific), anti-human SOX2 rat
monoclonal antibody (1:100; cat. no. 14-9811-82; Thermo Fisher
Scientific), anti-human type IV collagen rabbit polyclonal antibody
(1:200; cat. no. LB-0445; Life Science Laboratories, Inc.),
anti-p75NTR rabbit polyclonal antibody (1:200; cat. no. ANT-007;
Alomone Labs), and anti-human βB2-crystallin rabbit polyclonal
antibody (1:100; cat. no. ab252971; Abcam). Next, the cells were
incubated with an appropriate secondary antibody, Alexa Fluor
594-labeled anti-mouse IgG donkey antibody (1:500; cat. no.
A-21203; Thermo Fisher Scientific), Alexa Fluor 594-labeled
anti-rabbit IgG goat antibody (1:500; cat. no. A-11037; Thermo
Fisher Scientific), or Alexa Fluor 594-labeled anti-rat IgG goat
antibody (1:500; cat. no. A-11007; Thermo Fisher Scientific), for 1
h at 37˚C. DAPI (VECTASHIELD Mounting Medium with DAPI; cat. no.
H-1200; Vector Laboratories) was used for nuclear staining. The
immunostaining was evaluated using a fluorescence microscope (Power
BX-51; Olympus).
Statistical analysis
Each experiment was performed in triplicate and
repeated at least thrice. Data are presented as means ± standard
deviation (SD) and were analyzed using repeated measures analysis
of variance with Tukey's post hoc test. Statistical Package for
Social Science (SPSS) Statistics 24 (IBM Corporation) was used for
statistical analyses.
Results
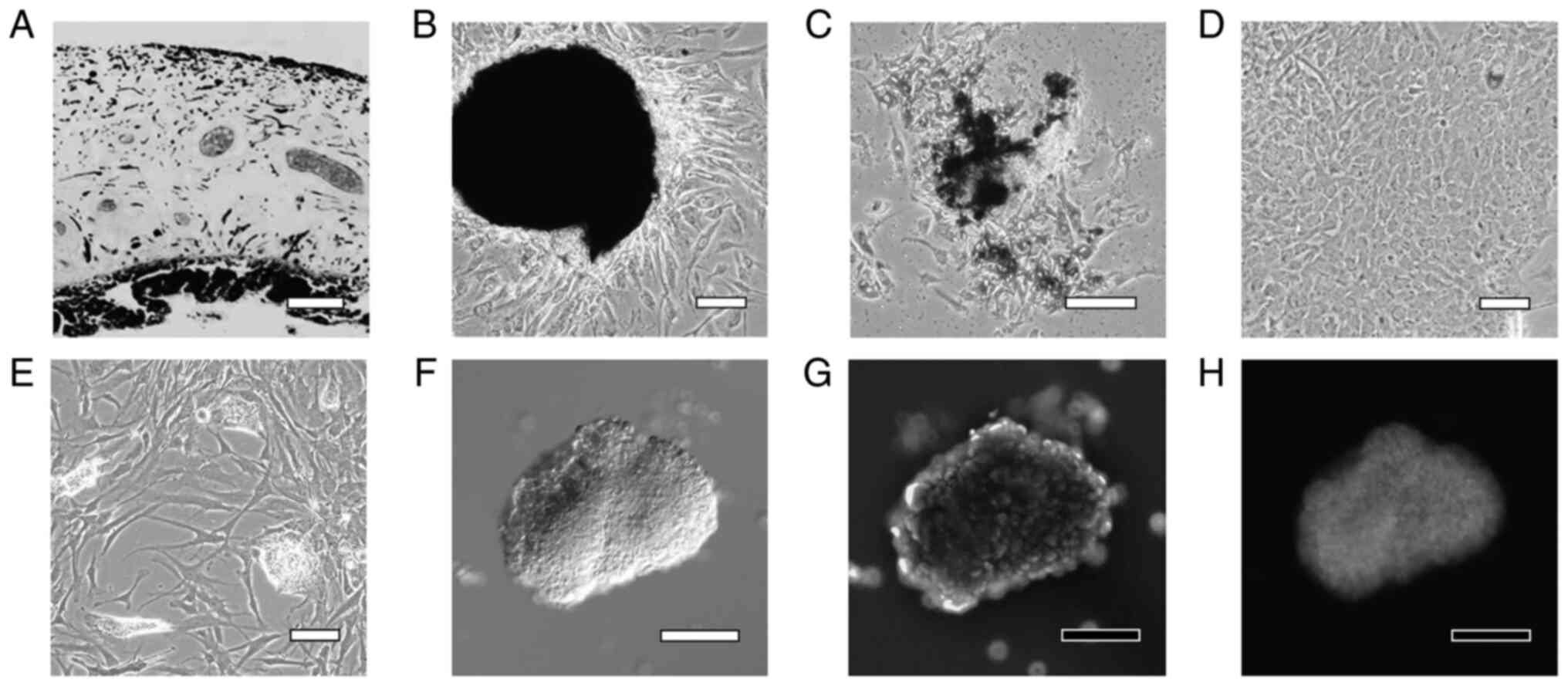
Primary culture and differentiation of
H-iris cells
Human iris tissue was enzymatically treated and
decomposed into small pieces (Fig.
2A). Cells proliferated from the tissue pieces that adhered to
dishes (Fig. 2B), and while some
of these cells contained a pigment in the cytoplasm, the pigment
was degranulated in several cells (Fig. 2C). On Day 7 of culture, the cells
became confluent and the cultures included very few pigmented cells
(Fig. 2D). After culturing in
STEP-4 medium for 3 weeks, the cultures contained small cell masses
(lentoid body-like masses) in which the cells were partially
aggregated (Fig. 2E). When the
small cell aggregates were collected using a pipette and
immunostained with an αA-crystallin antibody, numerous brightly
stained cells were observed (Fig.
2F-H).
Differentiation using the Ex.1
method
When iPS cells cultured in iPS medium (Fig. 3A) were cultured in STEP-1 medium
for 6 days, cells featuring short protrusions were observed
(Fig. 3B). After 12 days in STEP-2
medium, the cells were fully confluent and partially multilayered
(Fig. 3C), and after 17 days in
STEP-3 medium, the cells were further multilayered (Fig. 3D). On the last day of culture of
each step from STEP-0 to STEP-3, the cells were collected and total
RNA was extracted and reverse-transcribed to produce cDNA for PCR
analyses; our results showed that p75NTR mRNA expression
levels were significantly higher at STEP-1, -2, and -3 than at
STEP-0 (Fig. 3E). Moreover, at
STEP-3, the cultures included a few αA-crystallin-positive cell
populations, and the αA-crystallin-positive cells were negative for
SOX2 (Fig. 3F-I).
The expression of PAX6, SOX2, and αA-crystallin
proteins at each step was confirmed through immunostaining
performed under identical conditions. The expression of
αA-crystallin was strongest at STEP-3, but the expression was not
uniform, with certain cells expressing the protein more strongly
than others, and the expression tended to be stronger in aggregated
cells than in non-aggregated cells (Fig. 4). Fig.
4 examines the changes in protein expression of PAX6, SOX2, and
αA-crystallin during lens development in vivo at different
culture steps. Since iPS cells were used in this study, SOX2, one
of the markers of iPS cells, is strongly expressed in STEP-0.
However, by starting the induction of differentiation to lens, the
expression of SOX2 decreased once in STEP-1, but as the
differentiation STEP to lens progressed, the expression of SOX2
became strong again. On the other hand, PAX6 was slightly expressed
in STEP-1. In addition, αA-crystallin, a marker of lens protein,
was hardly detected until STEP-2, but was strongly detected in
STEP-3, indicating that the cells differentiated into lens
epithelial cells.
Differentiation using the Ex.2
method
The Ex.2 method was the same as the Ex.1 method
until the end of STEP-2, but in STEP-3, starting from 7 days after
initiation of the culture, the cells were cultured on a rotary
culture device (Fig. 5A). After 10
days of this rotational suspension culture, opaque cell aggregates
were formed (Fig. 5B). Sections of
the cell aggregates were prepared using the cell-block method, and
H&E staining revealed that the aggregates were surrounded by
one or two layers of cells, with the aggregate interior being
filled with cells distinct from the cells around the aggregates
(Fig. 5C). Moreover, the cells
forming the aggregates showed cytoplasmic staining for
αA-crystallin (Fig. 5D and
E). An illustration of the
developmental process of the lens is presented in Fig. 5F. Initially, a lens follicle
featuring a hollow center is formed, and then the interior of the
follicle is filled with cells extending from one direction on the
optic cup side.
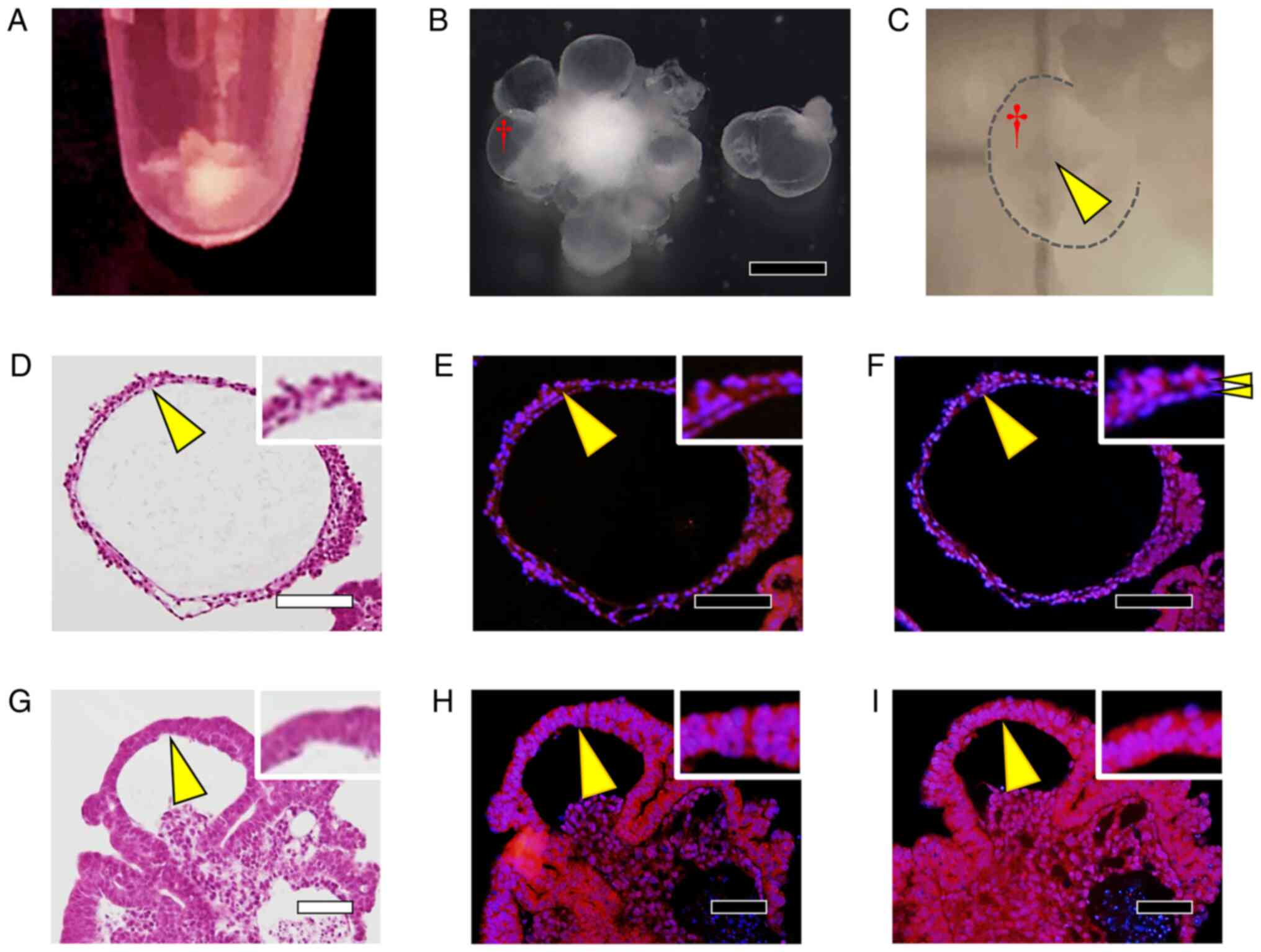
Differentiation by the Ex.3
method
In the Ex.3 method, cell aggregates were formed
starting from the STEP-1 stage, with differentiation occurring in
the cell aggregates, and several days after the start of culture in
STEP-3, numerous sac-like structures were observed around the cell
aggregates (Fig. 6A and B). The sac-like structures were
translucent (Fig. 6C), and H&E
staining of cell-block sections revealed that these structures were
formed by two layers of cell membranes, with the inner layer being
hollow (Fig. 6D). The two cell
layers showed identical staining for αA-crystallin (Fig. 6E), although the outer cells showed
stronger type IV collagen staining than did the inner cells
(Fig. 6F). Conversely, in H&E
staining of the cell aggregates, we observed that these structures
were formed by two layers of cells, and cells whose nuclei were
larger and cytoplasm was darkly stained with eosin were observed
compared to the sac-like structures (Fig. 6G). The cells whose cytoplasm was
darkly stained with eosin were also strongly positive for
αA-crystallin (Fig. 6H) and type
IV collagen (Fig. 6I) in both the
inner and outer layers of the two-layer structure.
Differentiation using the Ex.4
method
In the Ex.4 method, cells were cultured using the
static-suspension method to allow the formation of cell-cell
junctions for differentiation. Subsequently, a step involving
rotational suspension culture, STEP-4, was included for an
additional 2 weeks. Expression analysis of five genes revealed that
the expression levels in STEP-3 and STEP-4 were significantly
higher than the changes between STEP-0 and STEP-1 and STEP-2
(Fig. 7A-E). The expression levels
of p75NTR and type IV collagen were significantly higher in
STEP-3 than in STEP-0, with the expression levels of the two genes
decreasing in STEP-1 as compared to the level in STEP-0 but
increasing thereafter. The expression levels of SOX2 and
PAX6 increased during ocular differentiation, and in the
Ex.4 method, the expression levels of these genes were
significantly increased in STEP-3 and STEP-4.
Visual examination of cell aggregates obtained at
the end of culture (Fig. 8A)
revealed that the aggregates were milky-white and opaque, although
the opacity was lighter in certain areas than in others. The
results of H&E staining of sectioned specimens further showed
that vacuoles formed inside the cell aggregates (Fig. 8B). Moreover, the surface of the
cell aggregates was stratified, as illustrated in Fig. 8C showing an enlarged view of Area 1
from Fig. 8B, and these cells were
positive for SOX2 (Fig. 8D),
p75NTR (Fig. 8E), αA-crystallin
(Fig. 8F), and type IV collagen
(Fig. 8G). Conversely, in the
region that is marked as Area 2 in Fig. 8B and enlarged in Fig. 8H, the cells on the surface of the
aggregates transitioned from multilayers to monolayers, and the
monolayers were negative for SOX2 (Fig. 8I); furthermore, in this region,
p75NTR expression was decreased (Fig.
8J), and αA-crystallin staining was not detected (Fig. 8F), but positive staining for type
IV collagen was observed (Fig.
8G).
Differentiation using the Ex.5 (SEAM)
method
H-iris iPS cells (Fig.
9A) were cultured continuously for 4 weeks in a differentiation
medium of the same composition. At the end of culture, small cell
aggregates were observed at the boundary between the 2nd and 3rd
zones (Fig. 9B), and these
aggregates were positive for αA-crystallin (Fig. 9C). The cell aggregates were
carefully harvested using a pipette and cultured using the static-
and rotational-suspension methods. H&E staining of sections of
the cell aggregates revealed that the cells were arranged in
concentric circles, and in the cells in the interior of the
aggregates, the cytoplasm was uniformly stained with eosin
(Fig. 9D). αA-crystallin staining
was detected in the cells surrounding the aggregates and in the
inner cells whose cytoplasm was stained with eosin (Fig. 9E), and βB2-crystallin staining was
slightly stronger in the inner cells than in the cells around the
aggregates (Fig. 9F). Moreover,
the cells around and inside the cell aggregates were positive for
type IV collagen, and the inner cells whose cytoplasm was uniformly
stained with eosin also particularly stained strongly for type IV
collagen (Fig. 9G).
Discussion
In this study, we cultured human iris-derived tissue
cells and iPS cells using various culture methods and generated
three-dimensional cell aggregates expressing αA-crystallin, a
lens-specific protein.
The history of research on lens regeneration began
with lens regeneration in newts, which was discovered in the 1890s
(34,35). In newts, when the lens is removed,
the pigment in the iris degranulates and then the iris tissue
regenerates the lens. Lens regeneration in newts is unique, with
the lens being unfailingly regenerated from the dorsal iris.
Studies conducted using transgenic newts have shown that b-FGF and
Wnt play a major role in the regeneration of the lens, refuting the
notion that tissue stem cells exist only in the dorsal iris
(36), and lens regeneration has
also been shown to occur several times during the lifetime of a
newt (13,37). However, no study to date has
reported lens regeneration from the iris in mammals. We have been
conducting research in the field of regenerative medicine with a
focus on iris tissue (38-41).
Here, we found that lentoid body-like cell aggregates expressing a
very small but lens-specific protein were formed in cultures of
cells derived from human iris tissue. We previously reported that
cultured H-iris cells included cells positive for tissue stem cell
markers (CD271 and p75NTR) (26),
and p75NTR is also a tissue stem cell marker in the lens (42). Our method of culturing iris tissue
can also be used to culture certain cells that are positive for
tissue stem cell markers, and this is the first report of PECs in
human iris tissue degranulating and differentiating into small cell
aggregates (lentoid body-like cell masses) expressing
αA-crystallin, a marker for LECs.
First, we cultured H-iris iPS cells to induce their
differentiation into lens epithelial progenitor cells based on a
report that ES cells can differentiate into these progenitor cells
(29). As compared to the iPS
cells at STEP-0, the progressively differentiating cells showed
increasing mRNA expression of p75NTR, a marker of lens
epithelial stem cells. Moreover, PAX6 protein expression increased
during STEP-1, the stage at which p75NTR mRNA expression
started to increase, whereas SOX2 protein expression decreased once
during STEP-1 and increased again during STEP-3. The association
between PAX6 and SOX2 is critical because the two
have been reported to act in conjunction to activate the expression
of δ-crystallin and induce the development of lens placodes
(1). Cells expressing
αA-crystallin protein were observed during STEP-3, where
crystallin-positive cells were aggregated and not all cells
expressed αA-crystallin. Cell-cell interactions could play a
crucial role in lens differentiation by inducing the formation of
cell aggregates.
The maturation of cartilage cells has been reported
to be promoted by the application of a mechanical stimulation or
load to cells, and cellular aggregates generated using rotational
culture have been proposed to represent an essential component for
creating artificial cartilage through tissue engineering (43-47).
Here, from the middle of STEP-3, when rotational suspension culture
was performed as a mechanical stimulus (Ex.2), the cell aggregates
formed were surrounded by one or two layers of cells, and cells
elongating inward from certain directions were observed to fill the
interior of the aggregates. Our finding is similar to the observed
proliferation and migration of cells during lens formation during
development, and these cells expressed αA-crystallin. However, the
cell aggregates grew to <1 mm as the largest size.
Considering the aforementioned results, we next
cultured cells by employing the cell aggregates from STEP-1 and
subjecting them to tilt rotation and horizontal rotation (Ex.3).
Translucent sac-like structures formed by two layers of cell
membranes were observed. In the sac-like structures, type IV
collagen was strongly expressed outside the bilayer, mimicking the
type IV collagen-rich lens capsule present on the outer surface of
the lens (48-50).
However, because the process of development and growth of the lens
capsule remains unclear, embryological and cell-based studies on
the lens capsule are required.
As the next method, STEP-4 using LEC medium was
performed (Ex.4). All measured gene expression increased during
STEP-3 and further increased in STEP-4. The cell aggregates grew to
~2 mm in size, with the surrounding cells, including LECs, arranged
in the same manner as in the lens in certain areas, and a few
vacuolated areas were present in the interior.
Lastly, in Ex.5, the areas where LECs formed in
differentiated SEAM were collected and cultured. The SEAM method is
a two-dimensional adhesion-culture method, and the concentric SEAMs
formed by cells differentiating on their own through cell-cell
interaction without changing the medium conditions mimic the
development of the whole eye: the location of cells in different
zones shows lineages spanning the superficial ectoderm, lens,
neural retina, and retinal pigment epithelium of the eye. When
cells in the areas where superficial ectoderm differentiate under
the SEAM method are collected and further differentiated using the
air-lift method, a three-dimensional cell sheet expressing proteins
characteristic of the corneal and conjunctival epithelium is formed
(23,30,51).
Therefore, in the case of cells differentiated according to the
SEAM regions, differentiation is induced in a manner that is highly
distinct from the differentiation triggered previously in iPS cells
using other methods. A relatively uniform interior was detected in
the cell aggregates generated by collecting αA-crystallin-positive
cells formed using the SEAM method and then culturing them using
static- and rotational-suspension methods, and βB2-crystallin and
type IV collagen positivity was also detected. However, no lens
capsule was observed in these cell aggregates. It could be
challenging to completely control the directionality of cell
arrangement in cell aggregates without a lens capsule being present
as a basement membrane.
The lens nucleus is located at the center of the
lens, where organelles are lost during development. A recent study
on organelle degradation inside the lens reported that organelle
degradation by phospholipases of the PLAAT family leads to the
achievement of optimal transparency and refractive function of the
lens (4). Organelles affect light
scattering, and in a flow cytometer, a widely used research
instrument, the measurement is based on the principle that
laterally scattered light is affected by the size of the cell
nucleus and the presence of cell membranes and organelles (52,53).
We speculate that one of the reasons why the cell aggregates
produced in this study were not transparent is that intracellular
organelles remained and affected light scattering.
In the case of cataracts, a disease that causes
opacity of the lens, several artificial lenses (intraocular lenses;
IOLs) have been developed through micro-incisions and are being
used for clinical treatment. Regeneration of the lens in
vitro is highly intriguing as a basic research subject, but for
clinical application, the regeneration achieved must offer
advantages over current IOL-based treatments. Multifocal IOLs that
can focus at distinct distances are also being developed for
clinical use. In cataract surgery, the lens capsule is preserved
and only the opaque lens is emulsified using ultrasound and then
suctioned out. However, the IOLs currently used in clinical
practice are not perfect, and cataract surgery cannot enhance the
patient's ability to adjust the lens focus, particularly after
surgery. This is because the ciliary muscle connected to the lens
capsule is responsible for the focusing, and considering that the
ciliary body is also connected to the lens capsule (54), the in vitro regeneration of
the ciliary body is also related to the lens capsule and lens. We
aim to continue investigating the collective regeneration of the
lens, lens capsule, and ciliary body in vitro. We believe
that this can contribute to a finer focus adjustment after IOL
implantation if iPS cells can be used to regenerate weakened
ciliary bodies.
In conclusion, we have reported that H-iris cells
and iris tissue-derived iPS cells can be used to generate a variety
of three-dimensional cell aggregates expressing αA-crystallin, a
protein specific to the lens. However, all the cell aggregates
formed in this study were opaque, and we were thus unable to
regenerate a transparent lens. In the future, we will investigate
the degradation of organelles necessary to achieve transparency of
the interior of the generated cell aggregates expressing
lens-specific proteins by creating transgenic cells that induce the
disappearance of organelles, and we will conduct research on the
regeneration of transparent lenses. In addition, we aim to utilize
this cell aggregate model for various applications, such as
studying ciliary body regeneration by co-culture and creating an
in vitro cataract model that can evaluate the effects of
drugs and the effects of radiation exposure.
Acknowledgements
The authors would like to thank Ms. Chieko Nishikawa
(Fujita Health University) for help with experiments and Ms. Mari
Seto (Kanazawa Medical University) for English editing and
technical support.
Funding
Funding: This research was funded by MEXT/JSPS KAKENHI (grant
nos. 17K11495, 20K09838, and 20K09815).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
NH and NY designed the study. NH, NY and YK were
responsible for the data collection and manuscript writing. NH, NY,
YK, NN, SI and KI participated in the experiments. NH, YK, NN and
SI were responsible for data acquisition and analysis. NY and NN
were responsible for statistical analysis. YK and NN were
responsible for literature searches. NH, NY, YK and SI reviewed and
revised the manuscript. NY and KI confirm the authenticity of all
the raw data. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
This study was approved by the Ethics Review
Committee of Fujita Health University (approval no. 05-065, first
approval date: 21 December 2005, followed by continued ethical
approval; Aichi, Japan). The written informed consent was obtained
from all subjects. The experiment was carried out with the approval
of the Recombinant DNA Experiment Committee of Fujita Health
University (approval no. DP16055, approval date: 17 November 2016;
Aichi, Japan).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Kamachi Y, Uchikawa M, Tanouchi A, Sekido
R and Kondoh H: Pax6 and SOX2 form a co-DNA-binding partner complex
that regulates initiation of lens development. Genes Dev.
15:1272–1286. 2001.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Chow RL and Lang RA: Early eye development
in vertebrates. Annu Rev Cell Dev Biol. 17:255–296. 2001.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Iribarren R: Crystalline lens and
refractive development. Prog Retin Eye Res. 47:86–106.
2015.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Morishita H, Eguchi T, Tsukamoto S,
Sakamaki Y, Takahashi S, Saito C, Koyama-Honda I and Mizushima N:
Organelle degradation in the lens by PLAAT phospholipases. Nature.
592:634–638. 2021.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Kuszak JR, Zoltoski RK and Tiedemann CE:
Development of lens sutures. Int J Dev Biol. 48:889–902.
2004.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Yamamoto N, Tanikawa A and Horiguchi M:
Basic study of retinal stem/progenitor cell separation from mouse
iris tissue. Med Mol Morphol. 43:139–144. 2010.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Griep AE: Cell cycle regulation in the
developing lens. Semin Cell Dev Biol. 17:686–697. 2006.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Fujii N, Sakaue H, Sasaki H and Fujii N: A
rapid, comprehensive liquid chromatography-mass spectrometry
(LC-MS)-based survey of the Asp isomers in crystallins from human
cataract lenses. J Biol Chem. 287:39992–40002. 2012.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Delaye M and Tardieu A: Short-range order
of crystallin proteins accounts for eye lens transparency. Nature.
302:415–417. 1983.PubMed/NCBI View
Article : Google Scholar
|
|
10
|
Magami K, Hachiya N, Morikawa K, Fujii N
and Takata T: Isomerization of Asp is essential for assembly of
amyloid-like fibrils of αA-crystallin-derived peptide. PLoS One.
16(e0250277)2021.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Sprague-Piercy MA, Rocha MA, Kwok AO and
Martin RW: α-Crystallins in the vertebrate eye lens: Complex
oligomers and molecular chaperones. Annu Rev Phys Chem. 72:143–163.
2021.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Takata T, Murakami K, Toyama A and Fujii
N: Identification of isomeric aspartate residues in βB2-crystallin
from aged human lens. Biochim Biophys Acta Proteins Proteom.
1866:767–774. 2018.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Eguchi G, Eguchi Y, Nakamura K, Yadav MC,
Millán JL and Tsonis PA: Regenerative capacity in newts is not
altered by repeated regeneration and ageing. Nat Commun.
2(384)2011.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Barbosa-Sabanero K, Hoffmann A, Judge C,
Lightcap N, Tsonis PA and Del Rio-Tsonis K: Lens and retina
regeneration: New perspectives from model organisms. Biochem J.
447:321–334. 2012.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Gwon A: Lens regeneration in mammals: A
review. Surv Ophthalmol. 51:51–62. 2006.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Gwon A, Gruber LJ and Mantras C: Restoring
lens capsule integrity enhances lens regeneration in New Zealand
albino rabbits and cats. J Cataract Refract Surg. 19:735–746.
1993.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Gwon A, Gruber L, Mantras C and Cunanan C:
Lens regeneration in New Zealand albino rabbits after endocapsular
cataract extraction. Invest Ophthalmol Vis Sci. 34:2124–2129.
1993.PubMed/NCBI
|
|
18
|
Lin H, Ouyang H, Zhu J, Huang S, Liu Z,
Chen S, Cao G, Li G, Signer RA, Xu Y, et al: Lens regeneration
using endogenous stem cells with gain of visual function. Nature.
531:323–328. 2016.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Liu X, Zhang M and Liu Y, Challa P,
Gonzalez P and Liu Y: Proteomic analysis of regenerated rabbit
lenses reveal crystallin expression characteristic of adult
rabbits. Mol Vis. 14:2404–2412. 2008.PubMed/NCBI
|
|
20
|
Takahashi K, Tanabe K, Ohnuki M, Narita M,
Ichisaka T, Tomoda K and Yamanaka S: Induction of pluripotent stem
cells from adult human fibroblasts by defined factors. Cell.
131:861–872. 2007.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Chen P, Chen JZ, Shao CY, Li CY, Zhang YD,
Lu WJ, Fu Y, Gu P and Fan X: Treatment with retinoic acid and lens
epithelial cell-conditioned medium in vitro directed the
differentiation of pluripotent stem cells towards corneal
endothelial cell-like cells. Exp Ther Med. 9:351–360.
2015.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Qiu X, Yang J, Liu T, Jiang Y, Le Q and Lu
Y: Efficient generation of lens progenitor cells from cataract
patient-specific induced pluripotent stem cells. PLoS One.
7(e32612)2012.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Hayashi R, Ishikawa Y, Katori R, Sasamoto
Y, Taniwaki Y, Takayanagi H, Tsujikawa M, Sekiguchi K, Quantock AJ
and Nishida K: Coordinated generation of multiple ocular-like cell
lineages and fabrication of functional corneal epithelial cell
sheets from human iPS cells. Nat Protoc. 12:683–696.
2017.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Liu Z, Wang R, Lin H and Liu Y: Lens
regeneration in humans: Using regenerative potential for tissue
repairing. Ann Transl Med. 8(1544)2020.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Sun G, Asami M, Ohta H, Kosaka J and
Kosaka M: Retinal stem/progenitor properties of iris pigment
epithelial cells. Dev Biol. 289:243–252. 2006.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Yamamoto N, Hiramatsu N, Ohkuma M,
Hatsusaka N, Takeda S, Nagai N, Miyachi EI, Kondo M, Imaizumi K,
Horiguchi M, et al: Novel technique for retinal nerve cell
regeneration with electrophysiological functions using human
iris-derived iPS cells. Cells. 10(743)2021.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Drozd AM, Walczak MP, Piaskowski S,
Stoczynska-Fidelus E, Rieske P and Grzela DP: Generation of human
iPSCs from cells of fibroblastic and epithelial origin by means of
the oriP/EBNA-1 episomal reprogramming system. Stem Cell Res Ther.
6(122)2015.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Yamamoto N, Takeda S, Hatsusaka N,
Hiramatsu N, Nagai N, Deguchi S, Nakazawa Y, Takata T, Kodera S,
Hirata A, et al: Effect of a lens protein in low-temperature
culture of novel immortalized human lens epithelial cells
(iHLEC-NY2). Cells. 9(2670)2020.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Yang C, Yang Y, Brennan L, Bouhassira EE,
Kantorow M and Cvekl A: Efficient generation of lens progenitor
cells and lentoid bodies from human embryonic stem cells in
chemically defined conditions. FASEB J. 24:3274–3283.
2010.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Hayashi R, Ishikawa Y, Sasamoto Y, Katori
R, Nomura N, Ichikawa T, Araki S, Soma T, Kawasaki S, Sekiguchi K,
et al: Co-ordinated ocular development from human iPS cells and
recovery of corneal function. Nature. 531:376–380. 2016.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Hiramatsu N, Yamamoto N, Isogai S, Onouchi
T, Hirayama M, Maeda S, Ina T, Kondo M and Imaizumi K: An analysis
of monocytes and dendritic cells differentiated from human
peripheral blood monocyte-derived induced pluripotent stem cells.
Med Mol Morphol. 53:63–72. 2020.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Krogerus L and Kholova I: Cell block in
cytological diagnostics: Review of preparatory techniques. Acta
Cytol. 62:237–243. 2018.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Isogai S, Yamamoto N, Hiramatsu N, Goto Y,
Hayashi M, Kondo M and Imaizumi K: Preparation of induced
pluripotent stem cells using human peripheral blood monocytes. Cell
Reprogram. 20:347–355. 2018.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Henry JJ and Hamilton PW: Diverse
evolutionary origins and mechanisms of lens regeneration. Mol Biol
Evol. 35:1563–1575. 2018.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Papaconstantinou J: L.S. stone: Lens
regeneration-contributions to the establishment of an in vivo model
of transdifferentiation. J Exp Zool A Comp Exp Biol. 301:787–792.
2004.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Hayashi T, Mizuno N, Takada R, Takada S
and Kondoh H: Determinative role of Wnt signals in dorsal
iris-derived lens regeneration in newt eye. Mech Dev. 123:793–800.
2006.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Sousounis K, Qi F, Yadav MC, Millán JL,
Toyama F, Chiba C, Eguchi Y, Eguchi G and Tsonis PA: A robust
transcriptional program in newts undergoing multiple events of lens
regeneration throughout their lifespan. Elife.
4(e09594)2015.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Eguchi G, Abe SI and Watanabe K:
Differentiation of lens-like structures from newt iris epithelial
cells in vitro. Proc Natl Acad Sci USA. 71:5052–5056.
1974.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Kodama R and Eguchi G: From lens
regeneration in the newt to in-vitro transdifferentiation of
vertebrate pigmented epithelial cells. Semin Cell Biol. 6:143–149.
1995.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Kosaka M, Kodama R and Eguchi G: In vitro
culture system for iris-pigmented epithelial cells for molecular
analysis of transdifferentiation. Exp Cell Res. 245:245–251.
1998.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Abe T, Takeda Y, Yamada K, Akaishi K,
Tomita H, Sato M and Tamai M: Cytokine gene expression after
subretinal transplantation. Tohoku J Exp Med. 189:179–189.
1999.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Yamamoto N, Majima K and Marunouchi T: A
study of the proliferating activity in lens epithelium and the
identification of tissue-type stem cells. Med Mol Morphol.
41:83–91. 2008.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Furukawa KS, Suenaga H, Toita K, Numata A,
Tanaka J, Ushida T, Sakai Y and Tateishi T: Rapid and large-scale
formation of chondrocyte aggregates by rotational culture. Cell
Transplant. 12:475–479. 2003.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Nagai T, Furukawa KS, Sato M, Ushida T and
Mochida J: Characteristics of a scaffold-free articular chondrocyte
plate grown in rotational culture. Tissue Eng Part A. 14:1183–1193.
2008.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Duke PJ, Daane EL and Montufar-Solis D:
Studies of chondrogenesis in rotating systems. J Cell Biochem.
51:274–282. 1993.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Baker TL and Goodwin TJ: Three-dimensional
culture of bovine chondrocytes in rotating-wall vessels. In Vitro
Cell Dev Biol Anim. 33:358–365. 1997.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Marlovits S, Tichy B, Truppe M, Gruber D
and Schlegel W: Collagen expression in tissue engineered cartilage
of aged human articular chondrocytes in a rotating bioreactor. Int
J Artif Organs. 26:319–330. 2003.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Cummings CF and Hudson BG: Lens capsule as
a model to study type IV collagen. Connect Tissue Res. 55:8–12.
2014.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Matsuura-Hachiya Y, Arai KY, Muraguchi T,
Sasaki T and Nishiyama T: Type IV collagen aggregates promote
keratinocyte proliferation and formation of epidermal layer in
human skin equivalents. Exp Dermatol. 27:443–448. 2018.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Kelley PB, Sado Y and Duncan MK: Collagen
IV in the developing lens capsule. Matrix Biol. 21:415–423.
2002.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Nomi K, Hayashi R, Ishikawa Y, Kobayashi
Y, Katayama T, Quantock AJ and Nishida K: Generation of functional
conjunctival epithelium, including goblet cells, from human iPSCs.
Cell Rep. 34(108715)2021.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Barbiero G, Duranti F, Bonelli G, Amenta
JS and Baccino FM: Intracellular ionic variations in the apoptotic
death of L cells by inhibitors of cell cycle progression. Exp Cell
Res. 217:410–418. 1995.PubMed/NCBI View Article : Google Scholar
|
|
53
|
de Gann MP, Belaud-Rotureau MA, Voisin P,
Leducq N, Belloc F, Canioni P and Diolez P: Flow cytometric
analysis of mitochondrial activity in situ: Application to
acetylceramide-induced mitochondrial swelling and apoptosis.
Cytometry. 33:333–339. 1998.PubMed/NCBI
|
|
54
|
Kinoshita H, Suzuma K, Kaneko J, Mandai M,
Kitaoka T and Takahashi M: Induction of functional 3D ciliary
epithelium-like structure from mouse induced pluripotent stem
cells. Invest Ophthalmol Vis Sci. 57:153–161. 2016.PubMed/NCBI View Article : Google Scholar
|