Ion channels are pore-creating proteins that allow
the flow of inorganic ions through the cell membrane, plasma
membrane or intracellular organelles in a variety of tissues. They
can also provide a rapid diffusion pathway depending on the
electrochemical potential of the ions across the cell membrane.
Therefore, they can regulate the intracellular and extracellular
ion concentration involved in the establishment of membrane
potential, thus enabling them to play a notable role in several
physiological activities. Such basic physiological activities
include muscle contraction, protrusion transmission, action
potential transmission and cell secretion (1-6).
The three Nobel Prizes in physiology or medicine in
1963 and 1991 and in chemistry in 2003, revealed the necessary to
research the structure and function of ion channels (1). Ion channels are characterized by two
fundamental features, namely selectivity and gating (2). Therefore, depending on the
sensitivity of the gate to different stimuli, the ion channels can
be divided into voltage-, chemically- and mechanically gated
subgroups. The majority of ion channels, such as K+,
Na+, Ca2+, HCN and transient receptor
potential (TRP) channels, are similar in structure and form the
subgroup of voltage-gated channels since they may arise from the
same distant progenitor gene. Other ion channels such as
Cl-, aquaporins and junction proteins have different
structures compared with voltage-gated channels (1,2). The
function of ion channels is to respond to the opening and closing
of gating cues and to determine the types of ions that can pass
through. Additionally, ion channels consist of a ligand-binding and
ion permeation mechanism. Currently, the mechanisms underlying ion
permeation remains unclear (7,8).
Since several diseases are caused by ion channel mutations or other
dysfunctions, ion channels are considered major therapeutic targets
(9,10).
Proteinuria is a condition characterized by the
presence of protein in the urine at >150 mg/24 h or by positive
result from qualitative urinalysis testing. Proteinuria can be
divided into five categories: i) physiological proteinuria; ii)
glomerulinuria; iii) tubulminuria; iv) overload proteinuria; and v)
mixed proteinuria. The occurrence of proteinuria is closely
associated with impaired glomerular filtration membrane, which can
affect glomerular selective filtration function, thus allowing
several macromolecular substances, including proteins, to pass
through the filtration membrane, eventually promoting the
development of proteinuria (11).
The glomerular filtration membrane consists of glomerular capillary
endothelial cells (ECs), the glomerular basement membrane (GBM) and
renal capsule visceral layer epithelial cells (podocytes) (12-14).
The outermost layer of the glomerular basement membrane, the
outermost podocyte foot processes and its inter slit diaphragm (SD;
fissure membrane) are the main barriers of the glomerular
filtration membrane, as such they are key points of weakness in the
development of proteinuria (12-14).
The SD is a thin film between the adjacent bipedal processes on the
epithelial side of the GBM and is an isoaperture zipper-like
electron dense structure that forms the final part of the molecular
barrier and plays a significant role in preventing the efflux of
the majority of proteins (15).
When podocytes are damaged by several factors (such as genetic
abnormalities or infection) the expression of SD-related proteins
can decrease, leading to foot process fusion (a manifestation of
podocyte damage, which can increase glomerular permeability to
macromolecular substances) and GBM shedding, eventually resulting
in proteinuria. SD-associated proteins include nephrotic protein
(nephrin), podocyte cleft membrane protein (podocin),
CD2-associated protein and TRP cation channel protein (TRPC)
(16) and each protein plays a
different role. Nephrin maintains the structural strength of SD and
participates in the signal transmission of podocytes to regulate
the remodeling of podocyte cytoskeleton; podocin can form protein
complexes in the lipid microdomain of foot process membrane and
regulates SD filtration permeability through signal transduction;
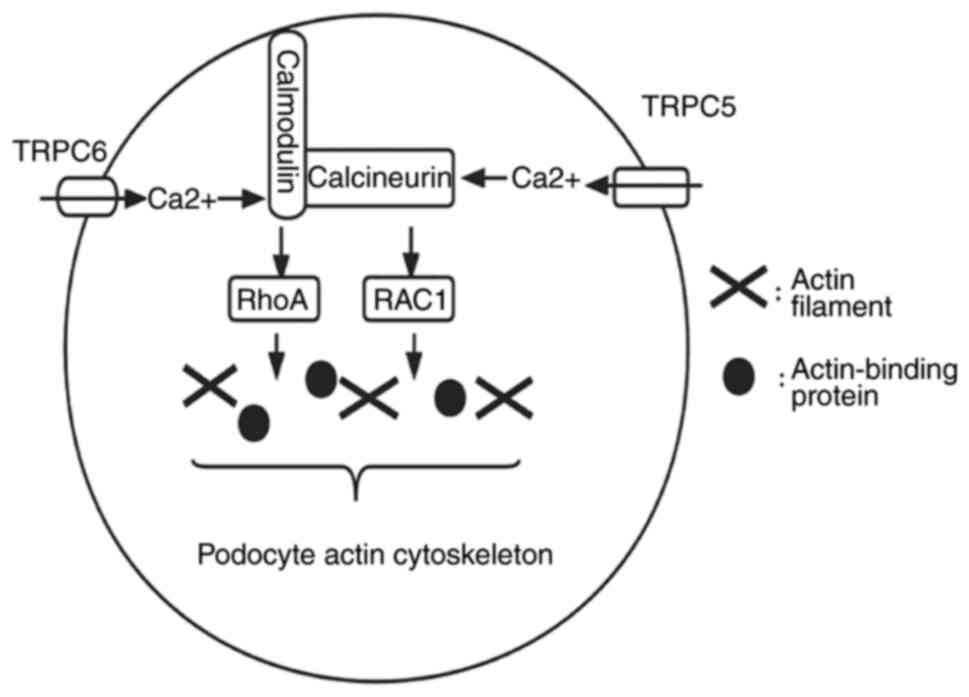
CD2AP is important for maintaining the integrity of the structure
and function of podocytes; and the role of TRPC is described in
Ca2+ channels (15,16).
In addition, proteinuria is one of the suggestive indicators of
kidney diseases and contributes to the diagnosis of these diseases.
Currently, studies have supported the association between the onset
of proteinuria and the activation or inhibition of ion channels
(8,12). Therefore, further understanding of
the mechanism underlying the action of ion channels and that of the
related ion transporters in proteinuria could advance targeted
therapy in patients with kidney disease-induced proteinuria.
TRP channels, a class of cationic channel proteins,
act as signal converters via altering membrane potential or the
intracellular concentration of Ca2+ (56,57).
In 1989, Montell and Rubin (58)
identified the TRP ion channels. TRP channels can be divided into
the following six subfamilies: i) TRPA (ankyrin); ii) TRPC
(canonical); iii) TRPM (melastatin); iv) TRPML (mucolipin); v) TRPP
(polycystin); and vi) TRPV (vanilloid) (59-61).
TRP channels can be activated through several pathways, such as
intracellular or extracellular transmitters, chemical stimulation
and osmotic pressure. Furthermore, the body can perceive the
changes of the external environment through the instantaneous
receptor potential channels in the surrounding environment, thus
protecting the body (62-67).
The current review article focused on the roles of TRPC and TRPV4
on proteinuria.
The TRPV4 receptor is expressed in the
cardiovascular system, including endothelial cells, cardiac
fibroblasts, VSMCs and perivascular nerves, but also in the
kidneys. This receptor can be activated by physical factors such as
high temperature and mechanical stimulation as well as by chemical
factors such as arachidonic acid (88-92).
Gualdani et al (93) showed
that fluid flow-induced mechanical force in proximal renal tubules
activates TRPV4 and promotes the endocytosis of albumin in proximal
renal tubular epithelial cells (Fig.
4), thus promoting albumin retention. Perineuria is also
significantly increased in TRPV4-depleted tubular cells in mice
when the glomerular filter permeability in the proximal tubules is
enhanced. Taken together, the TRPV4-mediated defects in endocytosis
may underlie proteinuria and therefore TRPV4 receptors can be
considered as promising targets for controlling proteinuria
(93).
Transporters are the other types of membrane
proteins that mediate the flow of ions through the cell membrane.
These proteins are different from ion channels. Pumps, the main
active transporters, use energy generated by ATP hydrolysis to
transport ions against electrochemical gradients, independent of
passive diffusion (131). In
addition, pumps are involved in several activities, such as
maintaining ion homeostasis by regulating the activity of pumps,
and require one or more proteins to maintain, including
Na+/K+-ATPases and
Na+/H+ exchanger proteins (131,132). Orlov et al (133) showed that proteinuria is
associated with abnormalities in the ion transporter
Na+/H+ exchange factor and
Na+/K+/2Cl- co-transporter in a
human model of primary hypertension. Hypertension and proteinuria
are the main features of preeclampsia (134). Graves (134) demonstrated that the above effect
is partially due to defects in Na+/K+-ATPases
or Na+ pumps. ClC-5 is the Cl-/H+
anti-transporter expressed in early proximal tubular endosomes
(135). When the function of
ClC-5 is impaired in endosomes, not only is the expression of
Na+/H+ exchange factors on the apical surface
of proximal tubules altered, but also reabsorption of low molecular
weight proteins and proximal tubular cell dysfunction are observed,
thus leading to albuminuria (135-137).
A study on a nephrotic syndrome rat model revealed that
amiloride-sensitive ENaCs are activated and the expression levels
of Na+ transporters are reduced in proximal renal
tubules, which may be due to the compensatory function (138). Furthermore, Gadau et al
(139) used an anti-Thy1
vasoproliferative glomerulonephritis rat model to evaluate the
association between the changes in Na+ and water
homeostasis and those in the renal tubular epithelium with
Na+ retention. The results showed that the abundance of
salt and water transporters (Na+/H+
exchanger-3, Na+/K+/2Cl-
co-transporter and aquaprin-1) in the proximal tubular brush border
membrane decreased, thus indicating that ion transporters are a
relevant mechanism of channel activation in glomerular diseases.
These findings suggest that there is an association between the
above mechanism and the occurrence and development of proteinuria.
In addition, de Seigneux et al (140) investigated the association
between proteinuria and phosphoremia in a nephrotic proteinuria
mouse model. The results showed that expression of
Na+/Na+ phosphate apical symporter is
abnormal in mice with proteinuria, while the albuminuria-induced
alterations on phosphate tubular handling are not associated with
the glomerular filtration rate. Another study by Shimizu et
al (141) exploring the
effects of N-acetylcysteine on kidney function and Na+
and water transporters in mice, showed that
Na+/K+/2Cl- co-transporter 2 and
aquaporin 2 are both upregulated and proteinuria is alleviated in
mice in that received the treatment. The above studies support an
association between proteinuria and ion transporters, thus
indicating that ion transporters could also be considered as
therapeutic targets for reducing proteinuria in patients.
Proteinuria is a significantly distinct determinant
of kidney disease progression and mortality. Therefore, unravelling
the mechanism of action of Na+, Ca2+,
Cl- and K+ channels and ion transporters in
proteinuria could provide novel insights into the identification of
novel targets for treating proteinuria and delaying kidney
diseases.
We thank Dr Yanqiang Wang (Department of Neurology
II, Affiliated Hospital of Weifang Medical University, Weifang,
China) for his instructive advice and useful suggestions and
providing help to finish the present review.
Funding: No funding was received.
Not applicable.
JL, XuL and NX wrote the review, collected
literature information and created figures. HH and XiL reviewed the
manuscript and proposed final revisions. All authors read and
approved the final manuscript. Data authentication is not
applicable.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Kondratskyi A, Kondratska K, Skryma R,
Klionsky DJ and Prevarskaya N: Ion channels in the regulation of
autophagy. Autophagy. 14:3–21. 2018.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Alexander SPH, Mathie A, Peters JA, Veale
EL, Striessnig J, Kelly E, Armstrong JF, Faccenda E, Harding SD,
Pawson AJ, et al: The concise guide to pharmacology 2019/20: Ion
channels. Br J Pharmacol. 176 (Suppl 1):S142–S228. 2019.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Roux B: Ion channels and ion selectivity.
Essays Biochem. 61:201–209. 2017.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Cheng J, Wen J, Wang N, Wang C, Xu Q and
Yang Y: Ion channels and vascular diseases. Arterioscler Thromb
Vasc Biol. 39:e146–e156. 2019.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Anderson KJ, Cormier RT and Scott PM: Role
of ion channels in gastrointestinal cancer. World J Gastroenterol.
25:5732–5772. 2019.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Armijo JA, Shushtarian M, Valdizan EM,
Cuadrado A, de las Cuevas I and Adín J: Ion channels and epilepsy.
Curr Pharm Des. 11:1975–2003. 2005.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Moiseenkova-Bell V, Delemotte L and Minor
DL Jr: Ion channels: Intersection of structure, function, and
pharmacology. J Mol Biol. 433(167102)2021.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Wang L and Yule DI: Differential
regulation of ion channels function by proteolysis. Biochim Biophys
Acta Mol Cell Res. 1865:1698–1706. 2018.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Catterall WA, Lenaeus MJ and Gamal El-Din
TM: Structure and pharmacology of voltage-gated sodium and calcium
channels. Annu Rev Pharmacol Toxicol. 60:133–154. 2020.PubMed/NCBI View Article : Google Scholar
|
|
10
|
De Logu F and Geppetti P: Ion channel
pharmacology for pain modulation. Handb Exp Pharmacol. 260:161–186.
2019.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Pallet N, Bastard JP, Claeyssens S,
Fellahi S, Delanaye P, Piéroni L and Caussé E: groupe de travail
SFBC, SFNDT, SNP. Proteinuria typing: How, why and for whom? Ann
Biol Clin (Paris). 77:13–25. 2019.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Barton M: Reversal of proteinuric renal
disease and the emerging role of endothelin. Nat Clin Pract
Nephrol. 4:490–501. 2008.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Nowak A and Serra AL: Assessment of
proteinuria. Praxis (Bern 1994). 102:797–802. 2013.PubMed/NCBI View Article : Google Scholar : (In German).
|
|
14
|
D'Amico G and Bazzi C: Pathophysiology of
proteinuria. Kidney Int. 63:809–825. 2003.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Menzel S and Moeller MJ: Role of the
podocyte in proteinuria. Pediatr Nephrol. 26:1775–1780.
2011.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Miner JH: Glomerular basement membrane
composition and the filtration barrier. Pediatr Nephrol.
26:1413–1417. 2011.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Kallen RG, Cohen SA and Barchi RL:
Structure, function and expression of voltage-dependent sodium
channels. Mol Neurobiol. 7:383–428. 1993.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Lee CH and Ruben PC: Interaction between
voltage-gated sodium channels and the neurotoxin, tetrodotoxin.
Channels (Austin). 2:407–412. 2008.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Hanukoglu I and Hanukoglu A: Epithelial
sodium channel (ENaC) family: Phylogeny, structure-function, tissue
distribution, and associated inherited diseases. Gene. 579:95–132.
2016.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Brunklaus A, Ellis R, Reavey E, Semsarian
C and Zuberi SM: Genotype phenotype associations across the
voltage-gated sodium channel family. J Med Genet. 51:650–658.
2014.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Soundararajan R, Pearce D, Hughey RP and
Kleyman TR: Role of epithelial sodium channels and their regulators
in hypertension. J Biol Chem. 285:30363–30369. 2010.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Büsst CJ: Blood pressure regulation via
the epithelial sodium channel: From gene to kidney and beyond. Clin
Exp Pharmacol Physiol. 40:495–503. 2013.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Zachar RM, Skjødt K, Marcussen N, Walter
S, Toft A, Nielsen MR, Jensen BL and Svenningsen P: The epithelial
sodium channel γ-subunit is processed proteolytically in human
kidney. J Am Soc Nephrol. 26:95–106. 2015.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Svenningsen P, Friis UG, Bistrup C, Buhl
KB, Jensen BL and Skøtt O: Physiological regulation of epithelial
sodium channel by proteolysis. Curr Opin Nephrol Hypertens.
20:529–533. 2011.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Ware AW, Rasulov SR, Cheung TT, Lott JS
and McDonald FJ: Membrane trafficking pathways regulating the
epithelial Na+ channel. Am J Physiol Renal Physiol.
318:F1–F13. 2020.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Bockenhauer D: Over- or underfill: Not all
nephrotic states are created equal. Pediatr Nephrol. 28:1153–1156.
2013.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Larionov A, Dahlke E, Kunke M, Zanon
Rodriguez L, Schiessl IM, Magnin JL, Kern U, Alli AA, Mollet G,
Schilling O, et al: Cathepsin B increases ENaC activity leading to
hypertension early in nephrotic syndrome. J Cell Mol Med.
23:6543–6553. 2019.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Fila M, Sassi A, Brideau G, Cheval L,
Morla L, Houillier P, Walter C, Gennaoui M, Collignon L, Keck M, et
al: A variant of ASIC2 mediates sodium retention in nephrotic
syndrome. JCI Insight. 6(e148588)2021.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Svenningsen P, Skøtt O and Jensen BL:
Proteinuric diseases with sodium retention: Is plasmin the link?
Clin Exp Pharmacol Physiol. 39:117–124. 2012.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Hinrichs GR, Jensen BL and Svenningsen P:
Mechanisms of sodium retention in nephrotic syndrome. Curr Opin
Nephrol Hypertens. 29:207–212. 2020.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Passero CJ, Hughey RP and Kleyman TR: New
role for plasmin in sodium homeostasis. Curr Opin Nephrol
Hypertens. 19:13–19. 2010.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Briet M and Schiffrin EL: Aldosterone:
Effects on the kidney and cardiovascular system. Nat Rev Nephrol.
6:261–273. 2010.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Shapovalov G, Skryma R and Prevarskaya N:
Calcium channels and prostate cancer. Recent Pat Anticancer Drug
Discov. 8:18–26. 2013.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Prevarskaya N, Ouadid-Ahidouch H, Skryma R
and Shuba Y: Remodelling of Ca2+ transport in cancer: How it
contributes to cancer hallmarks? Philos Trans R Soc Lond B Biol
Sci. 369(20130097)2014.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Mignen O, Thompson JL and Shuttleworth TJ:
Ca2+ selectivity and fatty acid specificity of the noncapacitative,
arachidonate-regulated Ca2+ (ARC) channels. J Biol Chem.
278:10174–10181. 2003.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Lewis RS: Store-operated calcium channels:
From function to structure and back again. Cold Spring Harb
Perspect Biol. 12(a035055)2020.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Zhou Y and Greka A: Calcium-permeable ion
channels in the kidney. Am J Physiol Renal Physiol.
310:F1157–F1167. 2016.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Patergnani S, Danese A, Bouhamida E,
Aguiari G, Previati M, Pinton P and Giorgi C: Various aspects of
calcium signaling in the regulation of apoptosis, autophagy, cell
proliferation, and cancer. Int J Mol Sci. 21(8323)2020.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Lou J, Yang X, Shan W, Jin Z, Ding J, Hu
Y, Liao Q, Du Q, Xie R and Xu J: Effects of calcium-permeable ion
channels on various digestive diseases in the regulation of
autophagy (review). Mol Med Rep. 24(680)2021.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Perez-Reyes E: Molecular physiology of
low-voltage-activated t-type calcium channels. Physiol Rev.
83:117–161. 2003.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Zamponi GW, Striessnig J, Koschak A and
Dolphin AC: The physiology, pathology, and pharmacology of
voltage-gated calcium channels and their future therapeutic
potential. Pharmacol Rev. 67:821–870. 2015.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Catterall WA: Voltage-gated calcium
channels. Cold Spring Harb Perspect Biol. 3(a003947)2011.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Sairaman A, Cardoso FC, Bispat A, Lewis
RJ, Duggan PJ and Tuck KL: Synthesis and evaluation of
aminobenzothiazoles as blockers of N- and T-type calcium channels.
Bioorg Med Chem. 26:3046–3059. 2018.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Tsunemi T, Saegusa H, Ishikawa K, Nagayama
S, Murakoshi T, Mizusawa H and Tanabe T: Novel Cav2.1 splice
variants isolated from Purkinje cells do not generate P-type Ca2+
current. J Biol Chem. 277:7214–7221. 2002.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Yamaguchi S, Okamura Y, Nagao T and
Adachi-Akahane S: Serine residue in the IIIS5-S6 linker of the
L-type Ca2+ channel alpha 1C subunit is the critical determinant of
the action of dihydropyridine Ca2+ channel agonists. J Biol Chem.
275:41504–41511. 2000.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Hansen PB: Functional and pharmacological
consequences of the distribution of voltage-gated calcium channels
in the renal blood vessels. Acta Physiol (Oxf). 207:690–699.
2013.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Ohta M, Sugawara S, Sato N, Kuriyama C,
Hoshino C and Kikuchi A: Effects of benidipine, a long-acting
T-type calcium channel blocker, on home blood pressure and renal
function in patients with essential hypertension: A retrospective,
‘real-world’ comparison with amlodipine. Clin Drug Investig.
29:739–746. 2009.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Mishima K, Maeshima A, Miya M, Sakurai N,
Ikeuchi H, Hiromura K and Nojima Y: Involvement of N-type Ca(2+)
channels in the fibrotic process of the kidney in rats. Am J
Physiol Renal Physiol. 304:F665–F673. 2013.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Tamargo J and Ruilope LM: Investigational
calcium channel blockers for the treatment of hypertension. Expert
Opin Investig Drugs. 25:1295–1309. 2016.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Khanna R, Yu J, Yang X, Moutal A,
Chefdeville A, Gokhale V, Shuja Z, Chew LA, Bellampalli SS, Luo S,
et al: Targeting the CaVα-CaVβ interaction yields an antagonist of
the N-type CaV2.2 channel with broad antinociceptive efficacy.
Pain. 160:1644–1661. 2019.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Ando K: L-/N-type calcium channel blockers
and proteinuria. Curr Hypertens Rev. 9:210–218. 2013.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Lei B, Nakano D, Fujisawa Y, Liu Y, Hitomi
H, Kobori H, Mori H, Masaki T, Asanuma K, Tomino Y and Nishiyama A:
N-type calcium channel inhibition with cilnidipine elicits
glomerular podocyte protection independent of sympathetic nerve
inhibition. J Pharmacol Sci. 119:359–367. 2012.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Fan YY, Kohno M, Nakano D, Ohsaki H,
Kobori H, Suwarni D, Ohashi N, Hitomi H, Asanuma K, Noma T, et al:
Cilnidipine suppresses podocyte injury and proteinuria in metabolic
syndrome rats: Possible involvement of N-type calcium channel in
podocyte. J Hypertens. 28:1034–1043. 2010.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Tamargo J: New calcium channel blockers
for the treatment of hypertension. Hipertens Riesgo Vasc. 34 (Suppl
2):S5–S8. 2017.PubMed/NCBI View Article : Google Scholar : (In Spanish).
|
|
55
|
Hansen PB, Poulsen CB, Walter S, Marcussen
N, Cribbs LL, Skøtt O and Jensen BL: Functional importance of L-
and P/Q-type voltage-gated calcium channels in human renal
vasculature. Hypertension. 58:464–470. 2011.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Nilius B and Owsianik G: The transient
receptor potential family of ion channels. Genome Biol.
12(218)2011.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Ramsey IS, Delling M and Clapham DE: An
introduction to TRP channels. Annu Rev Physiol. 68:619–647.
2006.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Montell C and Rubin GM: Molecular
characterization of the Drosophila trp locus: A putative integral
membrane protein required for phototransduction. Neuron.
2:1313–1323. 1989.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Gees M, Owsianik G, Nilius B and Voets T:
TRP channels. Compr Physiol. 2:563–608. 2012.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Caterina MJ and Pang Z: TRP channels in
skin biology and pathophysiology. Pharmaceuticals (Basel).
9(77)2016.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Li H: TRP channel classification. Adv Exp
Med Biol. 976:1–8. 2017.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Zhao Y, McVeigh BM and Moiseenkova-Bell
VY: Structural pharmacology of TRP channels. J Mol Biol.
433(166914)2021.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Nilius B: TRP channels in disease. Biochim
Biophys Acta. 1772:805–812. 2007.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Moran MM: TRP channels as potential drug
targets. Annu Rev Pharmacol Toxicol. 58:309–330. 2018.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Koivisto AP, Belvisi MG, Gaudet R and
Szallasi A: Advances in TRP channel drug discovery: From target
validation to clinical studies. Nat Rev Drug Discov. 21:41–59.
2022.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Feng YL, Chen H, Chen DQ, Vaziri ND, Su W,
Ma SX, Shang YQ, Mao JR, Yu XY, Zhang L, et al: Activated
NF-κB/Nrf2 and Wnt/β-catenin pathways are associated with lipid
metabolism in CKD patients with microalbuminuria and
macroalbuminuria. Biochim Biophys Acta Mol Basis Dis.
1865:2317–2332. 2019.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Voets T, Vriens J and Vennekens R:
Targeting TRP channels-valuable alternatives to combat pain, lower
urinary tract disorders, and type 2 diabetes? Trends Pharmacol Sci.
40:669–683. 2019.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Wang Q, Tian X, Wang Y, Wang Y, Li J, Zhao
T and Li P: Role of transient receptor potential canonical channel
6 (TRPC6) in diabetic kidney disease by regulating podocyte actin
cytoskeleton rearrangement. J Diabetes Res.
2020(6897390)2020.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Bacsa B, Tiapko O, Stockner T and
Groschner K: Mechanisms and significance of Ca2+ entry
through TRPC channels. Curr Opin Physiol. 17:25–33. 2020.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Möller CC, Flesche J and Reiser J:
Sensitizing the slit diaphragm with TRPC6 ion channels. J Am Soc
Nephrol. 20:950–953. 2009.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Möller CC, Wei C, Altintas MM, Li J, Greka
A, Ohse T, Pippin JW, Rastaldi MP, Wawersik S, Schiavi S, et al:
Induction of TRPC6 channel in acquired forms of proteinuric kidney
disease. J Am Soc Nephrol. 18:29–36. 2007.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Schaldecker T, Kim S, Tarabanis C, Tian D,
Hakroush S, Castonguay P, Ahn W, Wallentin H, Heid H, Hopkins CR,
et al: Inhibition of the TRPC5 ion channel protects the kidney
filter. J Clin Invest. 123:5298–5309. 2013.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Tian D, Jacobo SM, Billing D, Rozkalne A,
Gage SD, Anagnostou T, Pavenstädt H, Hsu HH, Schlondorff J, Ramos A
and Greka A: Antagonistic regulation of actin dynamics and cell
motility by TRPC5 and TRPC6 channels. Sci Signal.
3(ra77)2010.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Shalygin A, Shuyskiy LS, Bohovyk R,
Palygin O, Staruschenko A and Kaznacheyeva E: Cytoskeleton
rearrangements modulate TRPC6 channel activity in podocytes. Int J
Mol Sci. 22(4396)2021.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Schlondorff J: TRPC6 and kidney disease:
Sclerosing more than just glomeruli? Kidney Int. 91:773–775.
2017.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Schlöndorff JS and Pollak MR: TRPC6 in
glomerular health and disease: What we know and what we believe.
Semin Cell Dev Biol. 17:667–674. 2006.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Kim EY, Yazdizadeh Shotorbani P and Dryer
SE: Trpc6 inactivation confers protection in a model of severe
nephrosis in rats. J Mol Med (Berl). 96:631–644. 2018.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Hall G, Wang L and Spurney RF: TRPC
channels in proteinuric kidney diseases. Cells.
9(44)2019.PubMed/NCBI View Article : Google Scholar
|
|
79
|
van der Wijst J and Bindels RJM: Renal
physiology: TRPC5 inhibition to treat progressive kidney disease.
Nat Rev Nephrol. 14:145–146. 2018.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Walsh L, Reilly JF, Cornwall C, Gaich GA,
Gipson DS, Heerspink HJL, Johnson L, Trachtman H, Tuttle KR, Farag
YMK, et al: Safety and efficacy of GFB-887, a TRPC5 channel
inhibitor, in patients with focal segmental glomerulosclerosis,
treatment-resistant minimal change disease, or diabetic
nephropathy: TRACTION-2 trial design. Kidney Int Rep. 6:2575–2584.
2021.PubMed/NCBI View Article : Google Scholar
|
|
81
|
Reiser J, Polu KR, Möller CC, Kenlan P,
Altintas MM, Wei C, Faul C, Herbert S, Villegas I, Avila-Casado C,
et al: TRPC6 is a glomerular slit diaphragm-associated channel
required for normal renal function. Nat Genet. 37:739–744.
2005.PubMed/NCBI View
Article : Google Scholar
|
|
82
|
Wiggins RC: The spectrum of
podocytopathies: A unifying view of glomerular diseases. Kidney
Int. 71:1205–1214. 2007.PubMed/NCBI View Article : Google Scholar
|
|
83
|
Zhou Y, Castonguay P, Sidhom EH, Clark AR,
Dvela-Levitt M, Kim S, Sieber J, Wieder N, Jung JY, Andreeva S, et
al: A small-molecule inhibitor of TRPC5 ion channels suppresses
progressive kidney disease in animal models. Science.
358:1332–1336. 2017.PubMed/NCBI View Article : Google Scholar
|
|
84
|
Zhang H, Ding J, Fan Q and Liu S: TRPC6
up-regulation in Ang II-induced podocyte apoptosis might result
from ERK activation and NF-kappaB translocation. Exp Biol Med
(Maywood). 234:1029–1036. 2009.PubMed/NCBI View Article : Google Scholar
|
|
85
|
Wang Z, Wei X, Zhang Y, Ma X, Li B, Zhang
S, Du P, Zhang X and Yi F: NADPH oxidase-derived ROS contributes to
upregulation of TRPC6 expression in puromycin
aminonucleoside-induced podocyte injury. Cell Physiol Biochem.
24:619–626. 2009.PubMed/NCBI View Article : Google Scholar
|
|
86
|
Kistler AD, Singh G, Altintas MM, Yu H,
Fernandez IC, Gu C, Wilson C, Srivastava SK, Dietrich A, Walz K, et
al: Transient receptor potential channel 6 (TRPC6) protects
podocytes during complement-mediated glomerular disease. J Biol
Chem. 288:36598–36609. 2013.PubMed/NCBI View Article : Google Scholar
|
|
87
|
Zhou Y, Kim C, Pablo JLB, Zhang F, Jung
JY, Xiao L, Bazua-Valenti S, Emani M, Hopkins CR, Weins A and Greka
A: TRPC5 channel inhibition protects podocytes in
puromycin-aminonucleoside induced nephrosis models. Front Med
(Lausanne). 8(721865)2021.PubMed/NCBI View Article : Google Scholar
|
|
88
|
Randhawa PK and Jaggi AS: TRPV4 channels:
Physiological and pathological role in cardiovascular system. Basic
Res Cardiol. 110(54)2015.PubMed/NCBI View Article : Google Scholar
|
|
89
|
Everaerts W, Nilius B and Owsianik G: The
vanilloid transient receptor potential channel TRPV4: From
structure to disease. Prog Biophys Mol Biol. 103:2–17.
2010.PubMed/NCBI View Article : Google Scholar
|
|
90
|
Kassmann M, Harteneck C, Zhu Z, Nürnberg
B, Tepel M and Gollasch M: Transient receptor potential vanilloid 1
(TRPV1), TRPV4, and the kidney. Acta Physiol (Oxf). 207:546–564.
2013.PubMed/NCBI View Article : Google Scholar
|
|
91
|
Mannaa M, Markó L, Balogh A, Vigolo E,
N'diaye G, Kaßmann M, Michalick L, Weichelt U, Schmidt-Ott KM,
Liedtke WB, et al: Transient receptor potential vanilloid 4 channel
deficiency aggravates tubular damage after acute renal ischaemia
reperfusion. Sci Rep. 8(4878)2018.PubMed/NCBI View Article : Google Scholar
|
|
92
|
Karasawa T, Wang Q, Fu Y, Cohen DM and
Steyger PS: TRPV4 enhances the cellular uptake of aminoglycoside
antibiotics. J Cell Sci. 121:2871–2879. 2008.PubMed/NCBI View Article : Google Scholar
|
|
93
|
Gualdani R, Seghers F, Yerna X, Schakman
O, Tajeddine N, Achouri Y, Tissir F, Devuyst O and Gailly P:
Mechanical activation of TRPV4 channels controls albumin
reabsorption by proximal tubule cells. Sci Signal.
13(eabc6967)2020.PubMed/NCBI View Article : Google Scholar
|
|
94
|
Duran C, Thompson CH, Xiao Q and Hartzell
HC: Chloride channels: Often enigmatic, rarely predictable. Annu
Rev Physiol. 72:95–121. 2010.PubMed/NCBI View Article : Google Scholar
|
|
95
|
Xia J, Wang H, Li S, Wu Q, Sun L, Huang H
and Zeng M: Ion channels or aquaporins as novel molecular targets
in gastric cancer. Mol Cancer. 16(54)2017.PubMed/NCBI View Article : Google Scholar
|
|
96
|
Gururaja Rao S, Ponnalagu D, Patel NJ and
Singh H: Three decades of chloride intracellular channel proteins:
From organelle to organ physiology. Curr Protoc Pharmacol.
80:11.21.1–11.21.17. 2018.PubMed/NCBI View Article : Google Scholar
|
|
97
|
Berend K, van Hulsteijn LH and Gans RO:
Chloride: The queen of electrolytes? Eur J Intern Med. 23:203–211.
2012.PubMed/NCBI View Article : Google Scholar
|
|
98
|
Jentsch TJ, Stein V, Weinreich F and
Zdebik AA: Molecular structure and physiological function of
chloride channels. Physiol Rev. 82:503–568. 2002.PubMed/NCBI View Article : Google Scholar
|
|
99
|
Suzuki M, Morita T and Iwamoto T:
Diversity of Cl(-) channels. Cell Mol Life Sci. 63:12–24.
2006.PubMed/NCBI View Article : Google Scholar
|
|
100
|
Scheel O, Zdebik AA, Lourdel S and Jentsch
TJ: Voltage-dependent electrogenic chloride/proton exchange by
endosomal CLC proteins. Nature. 436:424–427. 2005.PubMed/NCBI View Article : Google Scholar
|
|
101
|
Uchida S: In vivo role of CLC chloride
channels in the kidney. Am J Physiol Renal Physiol. 279:F802–F808.
2000.PubMed/NCBI View Article : Google Scholar
|
|
102
|
Schriever AM, Friedrich T, Pusch M and
Jentsch TJ: CLC chloride channels in Caenorhabditis elegans. J Biol
Chem. 274:34238–34244. 1999.PubMed/NCBI View Article : Google Scholar
|
|
103
|
Jentsch TJ and Pusch M: CLC chloride
channels and transporters: Structure, function, physiology, and
disease. Physiol Rev. 98:1493–1590. 2018.PubMed/NCBI View Article : Google Scholar
|
|
104
|
Hryciw DH, Ekberg J, Pollock CA and
Poronnik P: ClC-5: A chloride channel with multiple roles in renal
tubular albumin uptake. Int J Biochem Cell Biol. 38:1036–1042.
2006.PubMed/NCBI View Article : Google Scholar
|
|
105
|
Novarino G, Weinert S, Rickheit G and
Jentsch TJ: Endosomal chloride-proton exchange rather than chloride
conductance is crucial for renal endocytosis. Science.
328:1398–1401. 2010.PubMed/NCBI View Article : Google Scholar
|
|
106
|
Devuyst O and Luciani A: Chloride
transporters and receptor-mediated endocytosis in the renal
proximal tubule. J Physiol. 593:4151–4164. 2015.PubMed/NCBI View Article : Google Scholar
|
|
107
|
Günther W, Piwon N and Jentsch TJ: The
ClC-5 chloride channel knock-out mouse-an animal model for Dent's
disease. Pflugers Arch. 445:456–462. 2003.PubMed/NCBI View Article : Google Scholar
|
|
108
|
Faria D, Rock JR, Romao AM, Schweda F,
Bandulik S, Witzgall R, Schlatter E, Heitzmann D, Pavenstädt H,
Herrmann E, et al: The calcium-activated chloride channel Anoctamin
1 contributes to the regulation of renal function. Kidney Int.
85:1369–1381. 2014.PubMed/NCBI View Article : Google Scholar
|
|
109
|
Yang YD, Cho H, Koo JY, Tak MH, Cho Y,
Shim WS, Park SP, Lee J, Lee B, Kim BM, et al: TMEM16A confers
receptor-activated calcium-dependent chloride conductance. Nature.
455:1210–1215. 2008.PubMed/NCBI View Article : Google Scholar
|
|
110
|
González C, Baez-Nieto D, Valencia I,
Oyarzún I, Rojas P, Naranjo D and Latorre R: K(+) channels:
Function-structural overview. Compr Physiol. 2:2087–2149.
2012.PubMed/NCBI View Article : Google Scholar
|
|
111
|
Sigworth FJ: Potassium channel mechanics.
Neuron. 32:555–556. 2001.PubMed/NCBI View Article : Google Scholar
|
|
112
|
Gulbis JM and Doyle DA: Potassium channel
structures: Do they conform? Curr Opin Struct Biol. 14:440–446.
2004.PubMed/NCBI View Article : Google Scholar
|
|
113
|
Kuang Q, Purhonen P and Hebert H:
Structure of potassium channels. Cell Mol Life Sci. 72:3677–3693.
2015.PubMed/NCBI View Article : Google Scholar
|
|
114
|
Noma A: ATP-regulated K+ channels in
cardiac muscle. Nature. 305:147–148. 1983.PubMed/NCBI View Article : Google Scholar
|
|
115
|
Ashcroft FM and Rorsman P: K(ATP) channels
and islet hormone secretion: New insights and controversies. Nat
Rev Endocrinol. 9:660–669. 2013.PubMed/NCBI View Article : Google Scholar
|
|
116
|
Rorsman P, Ramracheya R, Rorsman NJ and
Zhang Q: ATP-regulated potassium channels and voltage-gated calcium
channels in pancreatic alpha and beta cells: Similar functions but
reciprocal effects on secretion. Diabetologia. 57:1749–1761.
2014.PubMed/NCBI View Article : Google Scholar
|
|
117
|
Tinker A, Aziz Q, Li Y and Specterman M:
ATP-sensitive potassium channels and their physiological and
pathophysiological roles. Compr Physiol. 8:1463–1511.
2018.PubMed/NCBI View Article : Google Scholar
|
|
118
|
Cole WC, Chen TT and Clément-Chomienne O:
Myogenic regulation of arterial diameter: Role of potassium
channels with a focus on delayed rectifier potassium current. Can J
Physiol Pharmacol. 83:755–765. 2005.PubMed/NCBI View Article : Google Scholar
|
|
119
|
Jackson MB, Konnerth A and Augustine GJ:
Action potential broadening and frequency-dependent facilitation of
calcium signals in pituitary nerve terminals. Proc Natl Acad Sci
USA. 88:380–384. 1991.PubMed/NCBI View Article : Google Scholar
|
|
120
|
Guéguinou M, Chantôme A, Fromont G,
Bougnoux P, Vandier C and Potier-Cartereau M: KCa and Ca(2+)
channels: The complex thought. Biochim Biophys Acta.
1843:2322–2333. 2014.PubMed/NCBI View Article : Google Scholar
|
|
121
|
Sforna L, Megaro A, Pessia M, Franciolini
F and Catacuzzeno L: Structure, gating and basic functions of the
Ca2+-activated K channel of intermediate conductance. Curr
Neuropharmacol. 16:608–617. 2018.PubMed/NCBI View Article : Google Scholar
|
|
122
|
Walewska A, Kulawiak B, Szewczyk A and
Koprowski P: Mechanosensitivity of mitochondrial large-conductance
calcium-activated potassium channels. Biochim Biophys Acta
Bioenerg. 1859:797–805. 2018.PubMed/NCBI View Article : Google Scholar
|
|
123
|
Fezai M, Ahmed M, Hosseinzadeh Z and Lang
F: Up-regulation of the large-conductance Ca2+-activated K+ channel
by glycogen synthase kinase GSK3β. Cell Physiol Biochem.
39:1031–1039. 2016.PubMed/NCBI View Article : Google Scholar
|
|
124
|
Giebisch G: Potassium channels and the
kidney. Nephrologie. 21:223–228. 2000.PubMed/NCBI(In French).
|
|
125
|
Sorensen CM, Braunstein TH,
Holstein-Rathlou NH and Salomonsson M: Role of vascular potassium
channels in the regulation of renal hemodynamics. Am J Physiol
Renal Physiol. 302:F505–F518. 2012.PubMed/NCBI View Article : Google Scholar
|
|
126
|
Tamura Y, Tanabe K, Kitagawa W, Uchida S,
Schreiner GF, Johnson RJ and Nakagawa T: Nicorandil, a K(atp)
channel opener, alleviates chronic renal injury by targeting
podocytes and macrophages. Am J Physiol Renal Physiol.
303:F339–F349. 2012.PubMed/NCBI View Article : Google Scholar
|
|
127
|
Snijder PM, Frenay AR, Koning AM, Bachtler
M, Pasch A, Kwakernaak AJ, van den Berg E, Bos EM, Hillebrands JL,
Navis G, et al: Sodium thiosulfate attenuates angiotensin
II-induced hypertension, proteinuria and renal damage. Nitric
Oxide. 42:87–98. 2014.PubMed/NCBI View Article : Google Scholar
|
|
128
|
Hyodo T, Oda T, Kikuchi Y, Higashi K,
Kushiyama T, Yamamoto K, Yamada M, Suzuki S, Hokari R, Kinoshita M,
et al: Voltage-gated potassium channel Kv1.3 blocker as a potential
treatment for rat anti-glomerular basement membrane
glomerulonephritis. Am J Physiol Renal Physiol. 299:F1258–F1269.
2010.PubMed/NCBI View Article : Google Scholar
|
|
129
|
Huang C, Zhang L, Shi Y, Yi H, Zhao Y,
Chen J, Pollock CA and Chen XM: The KCa3.1 blocker TRAM34 reverses
renal damage in a mouse model of established diabetic nephropathy.
PLoS One. 13(e0192800)2018.PubMed/NCBI View Article : Google Scholar
|
|
130
|
Piwkowska A, Rogacka D, Audzeyenka I,
Kasztan M, Angielski S and Jankowski M: Insulin increases
glomerular filtration barrier permeability through PKGIα-dependent
mobilization of BKCa channels in cultured rat podocytes. Biochim
Biophys Acta. 1852:1599–1609. 2015.PubMed/NCBI View Article : Google Scholar
|
|
131
|
Neverisky DL and Abbott GW: Ion
channel-transporter interactions. Crit Rev Biochem Mol Biol.
51:257–267. 2015.PubMed/NCBI View Article : Google Scholar
|
|
132
|
Veiras LC, McFarlin BE, Ralph DL, Buncha
V, Prescott J, Shirvani BS, McDonough JC, Ha D, Giani J, Gurley SB,
et al: Electrolyte and transporter responses to angiotensin II
induced hypertension in female and male rats and mice. Acta Physiol
(Oxf). 229(e13448)2020.PubMed/NCBI View Article : Google Scholar
|
|
133
|
Orlov SN, Adragna NC, Adarichev VA and
Hamet P: Genetic and biochemical determinants of abnormal
monovalent ion transport in primary hypertension. Am J Physiol.
276:C511–C536. 1999.PubMed/NCBI View Article : Google Scholar
|
|
134
|
Graves SW: Sodium regulation, sodium pump
function and sodium pump inhibitors in uncomplicated pregnancy and
preeclampsia. Front Biosci. 12:2438–2446. 2007.PubMed/NCBI View Article : Google Scholar
|
|
135
|
Devuyst O, Jouret F, Auzanneau C and
Courtoy PJ: Chloride channels and endocytosis: New insights from
Dent's disease and ClC-5 knockout mice. Nephron Physiol.
99:p69–p73. 2005.PubMed/NCBI View Article : Google Scholar
|
|
136
|
Shipman KE and Weisz OA: Making a dent in
dent disease. Function (Oxf). 1(zqaa017)2020.PubMed/NCBI View Article : Google Scholar
|
|
137
|
Anglani F, Gianesello L, Beara-Lasic L and
Lieske J: Dent disease: A window into calcium and phosphate
transport. J Cell Mol Med. 23:7132–7142. 2019.PubMed/NCBI View Article : Google Scholar
|
|
138
|
Svenningsen P, Friis UG, Versland JB, Buhl
KB, Møller Frederiksen B, Andersen H, Zachar RM, Bistrup C, Skøtt
O, Jørgensen JS, et al: Mechanisms of renal NaCl retention in
proteinuric disease. Acta Physiol (Oxf). 207:536–545.
2013.PubMed/NCBI View Article : Google Scholar
|
|
139
|
Gadau J, Peters H, Kastner C, Kühn H,
Nieminen-Kelhä M, Khadzhynov D, Krämer S, Castrop H, Bachmann S and
Theilig F: Mechanisms of tubular volume retention in
immune-mediated glomerulonephritis. Kidney Int. 75:699–710.
2009.PubMed/NCBI View Article : Google Scholar
|
|
140
|
de Seigneux S, Wilhelm-Bals A and
Courbebaisse M: On the relationship between proteinuria and plasma
phosphate. Swiss Med Wkly. 147(w14509)2017.PubMed/NCBI View Article : Google Scholar
|
|
141
|
Shimizu MH, Volpini RA, de Bragança AC,
Campos R, Canale D, Sanches TR, Andrade L and Seguro AC:
N-acetylcysteine attenuates renal alterations induced by senescence
in the rat. Exp Gerontol. 48:298–303. 2013.PubMed/NCBI View Article : Google Scholar
|