Introduction
Multiple myeloma (MM) is an incurable malignant
plasma cell disease characterized by resistance to treatment
(1). Lenalidomide, the most widely
used immunomodulatory drug for the treatment of MM, has markedly
improved the survival and quality of life of patients with MM
(2-4).
However, a notable population of patients with MM develop
resistance to lenalidomide over time (5).
SET domain bifurcated histone lysine
methyltransferase 1 (SETDB1) is an epigenetic modifier primarily
involved in H3K9 methylation but can also mediate the methylation
of H3K27 and DNA (6). Previous
studies have implicated aberrant SETDB1 expression in the
development of various types of cancer, including head and neck,
lung, breast, ovarian, colorectal and hepatocellular cancer and
leukemia (7-13).
One of the primary mechanisms through which SETDB1 affects cancer
prognosis is the promotion of cell proliferation via AKT
methylation at lysine residues 64, 140 and 142, which results in
Thr-308 phosphorylation and increases AKT activity (14,15).
AKT dysregulation is associated with tumorigenesis and AKT
hyperactivity promotes chemotherapy resistance in various types of
cancer (16-18),
including MM (19-21).
Numerous mechanisms have been reported to contribute
to lenalidomide resistance in MM, including cereblon (CRBN)
downregulation, mutations in the RAS/MAPK pathway, TP53 and CRBN
cascade components (comprising Interferon regulatory factor 4 and
Ikaros family zinc finger 1), the overactivation of WNT/β-catenin
signaling along with CD44-triggered adhesion and MAPK cascade
overactivation (22-27).
However, the role and mechanisms of SETDB1 in lenalidomide
resistance remain unclear. Therefore, the present study aimed to
investigate the functional role of SETDB1 in myeloma cell
proliferation and MM cell resistance to lenalidomide, as well as
the potential underlying mechanisms.
Materials and methods
Cell lines and culture
The human MM cell lines, U266 and RPMI-8226 were
obtained from the American Type Culture Collection. The identity of
the cell lines was verified using short tandem repeat analysis and
cells were confirmed to be free from mycoplasma contamination. The
cells were cultured in DMEM with 10% FBS (both Gibco; Thermo Fisher
Scientific, Inc.) and 1% penicillin/streptomycin at 37˚C and 5%
CO2 in a humidified incubator.
Lentiviral vectors and infection
Human SETDB1 (NM_001145415; Origene) was amplified
and cloned into the lentiviral vector, pCDH-CMV-MCS-EF1-Pur
(Asia-Vector Biotechnology) and named SETDB1-OE. The empty
pCDH-CMV-MCS-EF1-Pur vectors served as the negative control
(SETDB1-NC). SETDB1 specific short hairpin RNA (shRNA) sequences
(Table I) were cloned into
plenti-shRNA-GFP-puro lentiviral vectors (Asia-Vector
Biotechnology), and the recombinant plasmids were named SETDB1-sh1,
SETDB1-sh2 and SETDB1-sh3. The non-targeted plenti-shRNA-GFP-puro
lentiviral vectors were used as the corresponding negative control
(shSETDB1-NC). 293T cells (Institute of Biochemistry and Cell
Biology, Shanghai, China) were transfected with lentiviral
constructs along with the second-generation packaging system
plasmid psPAX2 (cat. no. #12260; Addgene) and pMD2.G (cat. no.
#12259; Addgene) at a ratio of 3:3:1 (2.5 ug lentiviral plasmid +
2.5 ug psPAX2 + 0.83 ug pMD2.G), using Lipofectamine 2000
(Invitrogen, Carlsbad, CA, USA). At 48 h post-transfection, the
lentiviral supernatant was collected and filtered. To create stable
cell lines, MM cell lines were infected with lentiviral
supernatants at 37˚C for 24 h at a multiplicity of infection (MOI)
of 80, followed by the selection of stably transfected cells using
complete medium containing puromycin (1 µg/ml) for 4 days and then
maintained in 1 µg/ml puromycin.
 | Table IshRNA sequences of SETDB1. |
Table I
shRNA sequences of SETDB1.
| shRNA | Sense (5'-3') | Antisense
(5'-3') |
|---|
| SETDB1-shRNA1 |
CUGAUAGUCAGCAUGCGAA |
UUCGCAUGCUGACUAUCAG |
| SETDB1-shRNA2 |
UGGAGAAGAUGGAUUGUGU |
ACACAAUCCAUCUUCUCCA |
| SETDB1-shRNA3 |
AGGUGAAAUUUGACAACAA |
UUGUUGUCAAAUUUCACCU |
RNA extraction and reverse
transcription-quantitative PCR (RT-qPCR)
Total RNA from cells was extracted using
TRIzol® (Invitrogen; Thermo Fisher Scientific, Inc.).
The generation of cDNA was performed using PrimeScript RT reagent
kit (TransGen Biotech, Co., Ltd.) according to the manufacturer's
instructions. The qPCR was performed using SYBR® Premix
Ex Taq (Takara Bio, Inc.) with GAPDH as an endogenous control. The
thermocycling conditions were as follows: 95˚C for 3 min followed
by 45 cycles at 95˚C for 7 sec, 57˚C for 10 sec, 72˚C for 15 sec.
Relative quantification was calculated by the ΔΔCT method (28). The following primers were used for
RT-qPCR: SETDB-1 forward, 5'-taagacttggcacaaaggcac-3' and reverse,
5'-tccccgacagtagactctttc-3' and GAPDH forward,
5'-ggagcgagatccctccaaaat-3' and reverse,
5'-ggctgttgtcatacttctcatgg-3'.
Apoptosis analysis
To measure apoptosis, Treated MM cells were stained
with Annexin V-FITC and PI and assessed using a flow cytometer
(CytExpert software version 2.1; CytoFLEX flow cytometer; Beckman
Coulter, Inc.). Briefly, cells were rinsed twice with cold PBS and
centrifuged at 300 x g for 5 min at 4˚C. The cells were
re-suspended at 1x106 cells/ml in 250 µl 1X binding
buffer (BD Biosciences). A total of 100 µl suspension was placed in
a 5 ml tube and stained using 5 µl Annexin V-FITC and 10 µl PI for
15 min at room temperature in the dark. The proportion of apoptotic
cells including early and late apoptotic cells was then determined
using a flow cytometer.
Analysis of resistance to
lenalidomide
The drug resistance of MM cells was assessed using a
Cell Counting Kit-8 assay (CCK-8; BBI Life Sciences Corporation).
Briefly, MM cells were suspended at 1x105 cells/ml and
seeded in 96-well plates. The cells were then treated for 1 h with
lenalidomide at 0.00, 0.01, 0.10, 1.00, 5.00, 10.00, 100.00 and
1,000.00 µM at 37˚C. Subsequently, 10 µl CCK-8 reagent was added to
each well and the cells were incubated for 1 h at 37˚C, followed by
an absorbance reading at 450 nm using a microplate reader
(Biotek).
Western blot analysis
Proteins were extracted using RIPA lysis buffer
(cat. no. P0013B; Beyotime Institute of Biotechnology), and 20 µg
protein were loaded into each well. The quantity of protein was
determined by the BCA protein determination kit (cat. no. P0010;
Beyotime Institute of Biotechnology). Protein samples were
separated on 12% gels using SDS-PAGE and transferred onto
nitrocellulose membranes (Bio-Rad Laboratories, Inc.). The
membranes were blocked with 5% non-fat milk for 30 min at room
temperature and then inoculated with primary antibodies at 4˚C
overnight. The primary antibodies were as follows: AKT (1:1,000;
cat. no. #4685; Cell Signaling Technology, Inc.), N-cadherin
(1:1,000; cat. no. 22018-1-AP; Proteintech Group, Inc.), vimentin
(1:1,000; cat. no. #5741; Cell Signaling Technology, Inc.),
phosphorylated (p)-PI3K (1:1,000; cat. no. #4228; Cell Signaling
Technology, Inc), GAPDH (1:20,000; cat. no. #5174; Cell Signaling
Technology, Inc.), PI3K (1:1,000; cat. no. #4292; Cell Signaling
Technology, Inc.) and p-AKT (1:1,000; cat. no. #4060S; Cell
Signaling Technology, Inc.). Then, anti-mouse-IgG-HRP-conjugated
(1:1,000; cat. no. #7076; Cell Signaling Technology, Inc.) or
anti-rabbit-IgG-HRP-conjugated antibodies (1:1,000; cat. no. #7074;
Cell Signaling Technology, Inc.) were used as secondary antibodies.
The membranes were incubated with secondary antibodies at 25˚C for
2 h. The protein signal was developed using ECL Plus (Pierce;
Thermo Fisher Scientific, Inc.). The protein quantitative analysis
was performed using ImageJ 1.48u software (National Institutes of
Health).
Analysis of Gene Expression Omnibus
(GEO) datasets
A GEO dataset (accession no. GSE136324) obtained
from the GEO database (ncbi.nlm.nih.gov/gds) was used to assess the
association between SETDB1 levels and survival of patients with MM.
Based on median values, 867 patient samples were classified into
SETDB1-low and SETDB1-high expression groups, and Kaplan Meier
analysis was performed using default parameters.
To evaluate whether SETDB1 was differentially
expressed between the lenalidomide-resistant and -sensitive MM
cells, a dataset (accession no. GSE165557) was downloaded from GEO
containing genomic information from three lenalidomide-resistant
and three lenalidomide-sensitive MM cell lines.
In addition, the publicly available microarray data
(accession no. GSE136324) (29)was
downloaded from GEO database and SETDB1 expression was set as a
numeric variable. The metric for ranking genes in gene set
enrichment analysis (GSEA) was determined using Spearman's
correlation; other parameters were left as default. The cut-off
values for the significance of outcomes were FDR<0.25 and
|NES|>1. GSEA was run with the R clusterProfiler package
(version 4.6.0, bioconductor.org/packages/release/bioc/html/clusterProfiler.html)
(30).
Inhibition of the PI3K/AKT signaling
pathway
LY294002 (cat. no. #9901; Cell Signaling Technology,
Inc.), a specific inhibitor of the PI3K/AKT pathway, were diluted
to a concentration of 10 µg/ml. MM cells were cultured with
LY294002 solution at 37˚C for 24 h and harvested for flow
cytometry, CCK-8 assay, RT-qPCR and Western blot analysis.
Statistical analysis
Each experiment was repeated three times.
Statistical analysis was performed using GraphPad Prism 8
(Dotmatics), SPSS (version 25.0; IBM Corp.) and R statistical
software (version 3.0.2) using the package Two Stage Hazard Rate
Comparison (version 0.1-6; cran.r-project.org/web/packages/TSHRC/TSHRC.pdf).
Continuous variables are presented as the mean ± SD. Two-tailed
paired t-tests were conducted for the comparison between
lenalidomide-sensitive and -resistant MM cell lines in the GEO
dataset (GSE165557), while two-tailed unpaired t-test was used to
compare the data between two groups obeying normal distribution and
homogeneity of variance. Survival was evaluated using Kaplan-Meier
analysis. Progression-free survival (PFS) and overall survival (OS)
were assessed using a stratified log-rank test, and cases with
late-stage crossover of survival curves were evaluated using a
two-stage test (31). Differences
between >2 groups were compared using one-way ANOVA followed by
post hoc Tukey's test. P<0.05 was considered to indicate a
statistically significant difference.
Results
Upregulation of SETDB1 predicts poor
prognosis of MM patients
GSE136324 dataset was downloaded from the GEO
database. Kaplan-Meier analysis revealed that compared with
patients with low SETDB1, patients with high SETDB1 levels had a
significantly worse PFS and OS (Fig.
1A and B). This indicated that
SETDB1 upregulation was an unfavorable risk factor for survival of
patients with MM.
SETDB1 was increased in
lenalidomide-resistant MM cell lines
SETDB1 expression was analyzed in
lenalidomide-sensitive and -resistant MM cell lines using a GEO
dataset (GSE165557). Compared with the lenalidomide-sensitive
cells, SETDB1 expression was significantly increased in the
lenalidomide-resistant cell lines (Fig. 1C). Taken together, these results
indicated that SETDB1 upregulation in lenalidomide-resistant MM was
associated with poor prognosis. Thus, increased SETDB1 levels may
promote lenalidomide resistance and disease progression in MM.
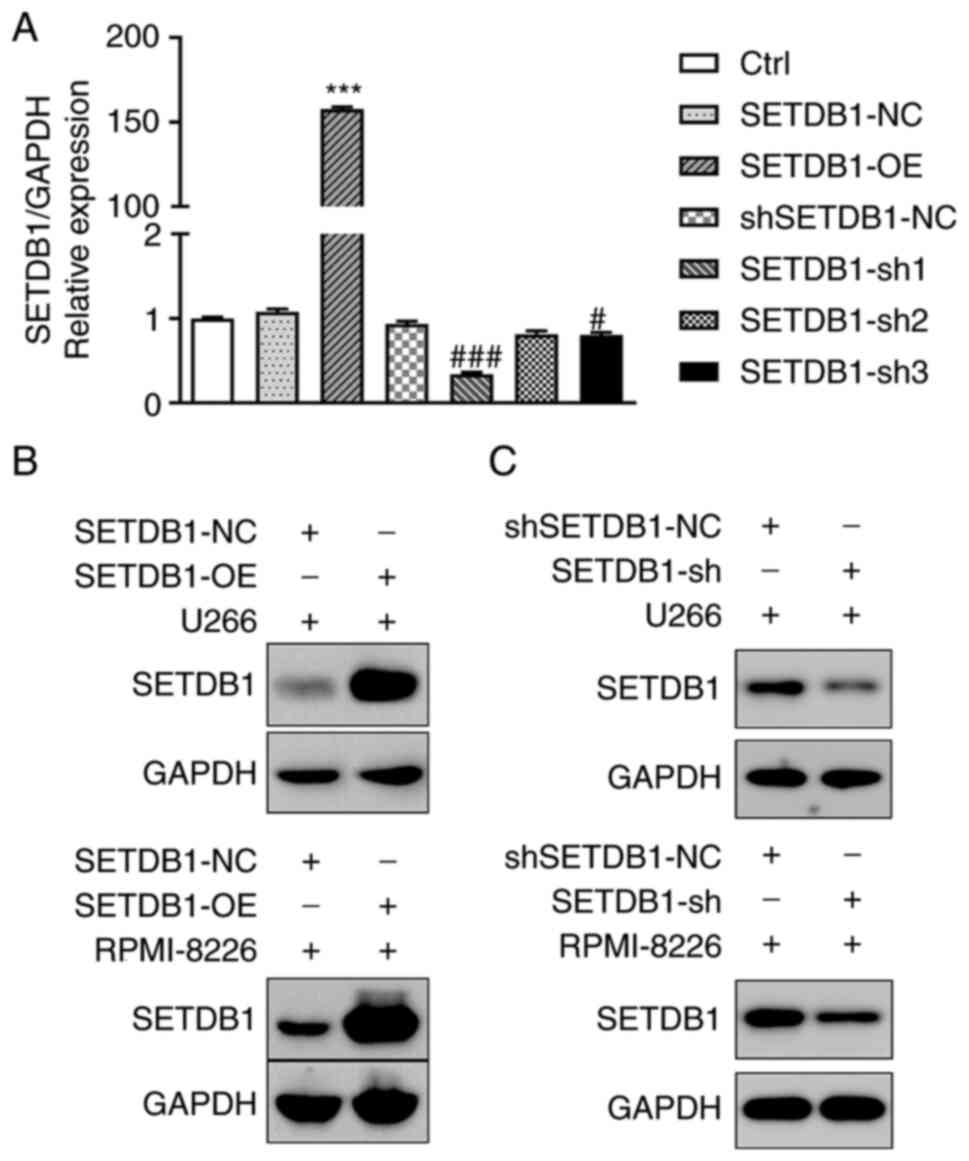
Construction of cell lines with stable
SETDB1 overexpression and knockdown
To determine the mechanisms through which SETDB1
contributes to drug resistance in MM cells and poor survival of
patients with MM, 293T cells were transfected with
SETDB1-overexpressing, SETDB1-shRNA (sh1, sh2 and sh3) and
respective NC lentivirus. Thus, stable SETDB1-OE, SETDB1-NC, SETDB1
knockdown (sh1, sh2 and sh3) and shSETDB1-NC MM cell lines were
obtained. Non-treated cells were used for control (Ctrl). RT-qPCR
revealed that SETDB1-OE demonstrated stable overexpression
efficiency, and SETDB1-sh1 demonstrated the greatest knockdown
efficiency (Fig. 2A). Results of
RT-qPCR were confirmed using western blot analysis. SETDB1 protein
expression was increased following transfection with SETDB1-OE in
MM cells (Fig. 2B) but decreased
following transfection with SETDB-sh1 (Fig. 2C). Thus, SETDB1-OE and SETDB1-sh1
(SETDB1-sh) were selected for use in subsequent experiments.
SETDB1 promotes apoptosis of MM
cells
The effect of SETDB1 overexpression or knockdown on
apoptosis was investigated using Annexin V-PI staining. In the U266
cells, compared with NC, apoptosis was significantly lower in the
SETDB1 overexpression group and significantly higher following
SETDB1 knockdown. Furthermore, similar observations were made in
the RPMI-8226 cells (Fig. 3A and
B). These results illustrated that
in MM cells, apoptosis was inhibited by SETDB1 overexpression and
enhanced by its knockdown.
SETDB1 expression is associated with
lenalidomide resistance in MM cells
The present study investigated the effects of
lenalidomide on the survival of MM cells in which SETDB1 was
overexpressed or silenced. MM cells were then treated with
lenalidomide for 1 h and cell proliferation was examined using
CCK-8 assay. IC50 value of lenalidomide was increased by
SETDB1 overexpression and decreased by SETDB1 knockdown (Fig. 3C and D). These results indicated that SETDB1
promoted lenalidomide resistance in MM cells.
SETDB1 expression is associated with
PI3K/AKT signaling and epithelial-mesenchymal transition (EMT)
activation in MM cells
GSEA of public microarray data indicated that genes
in the PI3K/AKT/mTOR signaling pathway were positively associated
with SETDB1 (Fig. 4A). Western
blot analysis illustrated that phosphorylation levels of PI3K and
AKT were significantly increased in the SETDB1 overexpression group
and significantly decreased in the SETDB1 knockdown group (Fig. 4B and C). Therefore, SETDB1 activation of
PI3K/AKT signaling by phosphorylating PI3K along with AKT may be
one of the mechanisms by which SETDB1 promotes MM progression. EMT
is a phase in cancer metastasis that is characterized by increased
expression of mesenchymal markers, such as N-cadherin and vimentin
(32). Therefore, to examine the
effect of SETDB1 on MM invasion, western blot analysis was used to
assess the levels of N-cadherin and vimentin. SETDB1 overexpression
significantly increased the levels of N-cadherin and vimentin in MM
cells, while SETDB1 knockdown significantly decreased these levels
(Fig. 4B and C). These results illustrated that SETDB1
upregulation promoted EMT in MM cells.
PI3K/AKT signaling is involved in
SETDB1-induced lenalidomide resistance and EMT in MM cells
To assess the role of PI3K/AKT signaling in the
SETDB1-trigerred lenalidomide resistance of MM cells, MM cells were
treated with LY294002, a specific inhibitor of PI3K/AKT signaling
(33). LY294002 significantly
increased the apoptosis of MM cells, while SETDB1 overexpression
significantly decreased apoptosis even following treatment with
LY294002 (Fig. 5A and B). LY294002 increased the sensitivity of
MM cells to lenalidomide, while SETDB1 overexpression counteracted
the effects of LY294002 (Fig. 5C).
Western blot analysis revealed that LY294002 significantly
decreased the levels of N-cadherin and vimentin and phosphorylation
of PI3K and AKT, however, SETDB1 overexpression partially offset
the effects of LY294002 (Fig. 5D).
Collectively, these results indicated that inhibition of PI3K/AKT
signaling promoted the apoptosis of MM cells, sensitized MM cells
to lenalidomide and inhibited EMT of MM cells, however, SETDB1
overexpression suppressed the effects of PI3K/AKT signaling
inhibition. Furthermore, these results demonstrated that SETDB1
promoted lenalidomide resistance and EMT in MM cells by increasing
phosphorylation of PI3K and AKT and thus activating PI3K/AKT
signaling.
 | Figure 5PI3K/AKT signaling and
epithelial-mesenchymal transition contribute to SETDB1-induced
lenalidomide resistance in multiple myeloma. (A) Flow cytometry
analysis of apoptosis of U266 and RPMI-8226 cells after treatment
with LY294002 and SETDB1 overexpression. (B) Statistical analysis
of apoptosis in each group. **P<0.01,
***P<0.001 vs. SETDB1-NC; ###P<0.001
vs. LY294002 + SETDB1-NC. (C) IC50 values of
lenalidomide were determined by Cell Counting Kit-8 assay. LY294002
significantly reduced the IC50 value of lenalidomide,
whereas SETDB1 overexpression attenuated this effect. (D) Western
blot analysis revealed that the expression of N-cadherin and
vimentin, as well as the phosphorylation of PI3K and AKT, were
inhibited by LY294002; these effects were counteracted by SETDB1
overexpression. ***P<0.001 vs. SETDB1-NC;
#P<0.05, ##P<0.01 and
###P<0.001 vs. LY294002 + SETDB1-NC. SETDB1, SET
domain bifurcated histone lysine methyltransferase 1; NC, negative
control; OE, overexpression; IC50, half maximal
inhibitory concentration; N-cad, N-cadherin; p,
phosphorylation. |
Discussion
MM is an incurable cancer of B cells that is defined
by the build-up of myeloma cells in the bone marrow, resulting in
bone damage, anemia, renal impairment and hypercalcemia (34). Lenalidomide is a second-generation
immunomodulatory drug approved for treatment of primary and
relapsed MM. Despite promising clinical activity, a number of
patients with MM do not respond to lenalidomide due to drug
resistance (35). Furthermore,
lenalidomide monotherapy in relapsed or refractory MM has only a
26% partial response rate, with a median PFS of 4.9 months and a
median OS of 23.2 months (36,37).
Thus, to improve the outcomes of patients with MM, it is necessary
to overcome lenalidomide resistance.
Epigenetic modifications contribute to abnormal gene
expression in cancer. SETDB1, a histone lysine methyltransferase,
is involved in methylation of DNA and the silencing of genes
(15). By suppressing tumor
suppressor genes, aberrant histone methylation of H3K9 by SETDB1
leads to carcinogenesis (38-40).
Thus, SETDB1 may be a promising therapeutic target.
In the present study, analysis of the GEO datasets
revealed that SETDB1 was upregulated in lenalidomide-resistant MM
cell lines and that patients with MM with high SETDB1 expression
had a worse PFS and OS compared with patients with low SETDB1
levels. These results indicated that SETDB1 affects prognosis of MM
and may be associated with lenalidomide resistance. Previous
studies have established that SETDB1 is highly expressed in various
types of cancer and affects prognosis (41-43),
which is consistent with the findings of the present study.
However, to the best of our knowledge, there are no reports that
indicate that SETDB1 is involved in drug resistance.
In the present study, to elucidate the mechanisms
underlying lenalidomide resistance in myeloma cells,
SETDB1-overexpressing and -knockdown plasmids were transfected into
U266 and RPMI-8226 myeloma cells. First, it was confirmed that
SETDB1 silencing markedly attenuated the proliferation of MM cells
and enhanced sensitivity to lenalidomide, whereas SETDB1
overexpression exerted the opposite effects. These results
indicated that while SETDB1 silencing may suppress cell
proliferation, induce apoptosis and sensitize MM cells to
lenalidomide, its overexpression may promote lenalidomide
resistance in MM cells.
GSEA revealed that SETDB1 was associated with the
PI3K/AKT/mTOR signaling pathway. PI3K/AKT signaling is activated by
binding of cytokines, including insulin-like growth factor, IL-6
and VEGF, to cell surface receptors, which triggers phosphorylation
and activation of AKT. AKT activates proliferation and
anti-apoptosis via mTOR-dependent and -independent pathways
(44-46).
The PI3K/AKT signaling pathway has been reported to promote the
proliferation and the survival of myeloma cells (47-49).
This pathway is necessary for MM cell proliferation and drug
resistance, including chemotherapy, radiotherapy and bortezomib
treatment (21,50).
EMT is a step in carcinogenesis that mediates cancer
cell mobility, invasion and resistance to apoptotic stimuli
(51). There is increasing
evidence to indicate that EMT activation promotes cancer drug
resistance (52-54).
In MM, early myeloma recurrence is associated with EMT (55). It has been demonstrated that EMT
modulation via PI3K/AKT signaling promotes MM progression (56). In the present study, SETDB1
promoted the survival and proliferation of MM cells via EMT and
PI3K/AKT pathway activation, and similar findings have been
previously reported in other tumors (9,57),
but this is the first time it has been confirmed in MM cells.
Moreover, we found that SETDB1 induced lenalidomide resistance in
multiple myeloma cells via epithelial-mesenchymal transition and
PI3K/AKT pathway activation, which had not been previously
reported. Furthermore, these findings demonstrated that SETDB1
functioned via the activation of the PI3K/AKT cascade and EMT.
In conclusion, the present study demonstrated that
SETDB1 expression was elevated in lenalidomide-resistant MM cells
and affected prognosis of patients with MM. The functional role of
SETDB1 in MM cell proliferation and lenalidomide resistance was
partly mediated by activation of EMT and PI3K/AKT signaling. Thus,
SETDB1 may be a potential therapeutic target for MM.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by the Science and
Technology Development Foundation project of Jiangsu University
(grant no. JLY2021188) and Taizhou Science and Technology Support
Project (grant no. TS202226).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HY conceived the experiments and wrote the
manuscript. YY, YX and YL performed the experiments. XQ, YY, YD, YZ
and FH analyzed the data and prepared the figures. XQ and YY
confirm the authenticity of all the raw data. All authors reviewed
the manuscript. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Palumbo A and Anderson K: Multiple
myeloma. N Engl J Med. 364:1046–1060. 2011.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Benboubker L, Dimopoulos MA, Dispenzieri
A, Catalano J, Belch AR, Cavo M, Pinto A, Weisel K, Ludwig H,
Bahlis N, et al: Lenalidomide and dexamethasone in
transplant-ineligible patients with myeloma. N Eng J Med.
371:906–917. 2014.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Palumbo A, Hajek R, Delforge M, Kropff M,
Petrucci MT, Catalano J, Gisslinger H, Wiktor-Jędrzejczak W,
Zodelava M, Weisel K, et al: Continuous lenalidomide treatment for
newly diagnosed multiple myeloma. N Engl J Med. 366:1759–1769.
2012.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Zonder JA, Crowley J, Hussein MA, Bolejack
V, Moore DF Sr, Whittenberger BF, Abidi MH, Durie BGM and Barlogie
B: Lenalidomide and high-dose dexamethasone compared with
dexamethasone as initial therapy for multiple myeloma: A randomized
Southwest oncology group trial (S0232). Blood. 116:5838–5841.
2010.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Ng YLD, Ramberger E, Bohl SR, Dolnik A,
Steinebach C, Conrad T, Müller S, Popp O, Kull M, Haj M, et al:
Proteomic profiling reveals CDK6 upregulation as a targetable
resistance mechanism for lenalidomide in multiple myeloma. Nat
Commun. 13(1009)2022.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Harte PJ, Wu W, Carrasquillo MM and Matera
AG: Assignment of a novel bifurcated SET domain gene, SETDB1, to
human chromosome band 1q21 by in situ hybridization and radiation
hybrids. Cytogenet Cell Genet. 84:83–86. 1999.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Özdaş S: Knockdown of SET domain,
Bifurcated 1 suppresses head and neck cancer cell viability and
wound-healing ability in vitro. Turk J Biol. 43:281–292.
2019.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Kang YK and Min B: SETDB1 overexpression
sets an intertumoral transcriptomic divergence in non-small cell
lung carcinoma. Front Genet. 11(573515)2020.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Yang W, Su Y, Hou C, Chen L, Zhou D, Ren
K, Zhou Z, Zhang R and Liu X: SETDB1 induces epithelial-mesenchymal
transition in breast carcinoma by directly binding with Snail
promoter. Oncol Rep. 41:1284–1292. 2019.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Wang W, Wang J, Zhang X and Liu G: Serum
circSETDB1 is a promising biomarker for predicting response to
platinum-taxane-combined chemotherapy and relapse in high-grade
serous ovarian cancer. Onco Targets Ther. 12:7451–7457.
2019.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Ropa J, Saha N, Chen Z, Serio J, Chen W,
Mellacheruvu D, Zhao L, Basrur V, Nesvizhskii AI and Muntean AG:
PAF1 complex interactions with SETDB1 mediate promoter H3K9
methylation and transcriptional repression of Hoxa9 and Meis1 in
acute myeloid leukemia. Oncotarget. 9:22123–22136. 2018.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Carvalho S, Freitas M, Antunes L,
Monteiro-Reis S, Vieira-Coimbra M, Tavares A, Paulino S, Videira
JF, Jerónimo C and Henrique R: Prognostic value of histone marks
H3K27me3 and H3K9me3 and modifying enzymes EZH2, SETDB1 and LSD-1
in colorectal cancer. J Cancer Res Clin Oncol. 144:2127–2137.
2018.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Wang C, Xia Z, Li Z, Ye F, Ji S, Lu C and
Zhang H: Expression of SET domain bifurcated histone lysine
methyltransferase 1 and its clinical prognostic significance in
hepatocellular carcinoma. J Clin Lab Anal.
36(e24090)2022.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Wang G, Long J, Gao Y, Zhang W, Han F, Xu
C, Sun L, Yang SC, Lan J, Hou Z, et al: SETDB1-mediated methylation
of Akt promotes its K63-linked ubiquitination and activation
leading to tumorigenesis. Nat Cell Biol. 21:214–225.
2019.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Guo J, Dai X, Laurent B, Zheng N, Gan W,
Zhang J, Guo A, Yuan M, Liu P, Asara JM, et al: AKT methylation by
SETDB1 promotes AKT kinase activity and oncogenic functions. Nat
Cell Biol. 21:226–237. 2019.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Kim SH, Juhnn YS and Song YS: Akt
involvement in paclitaxel chemoresistance of human ovarian cancer
cells. Ann N Y Acad Sci. 1095:82–89. 2007.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Guerrero-Zotano A, Mayer IA and Arteaga
CL: PI3K/AKT/mTOR: Role in breast cancer progression, drug
resistance, and treatment. Cancer Metastasis Rev. 35:515–524.
2016.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Shorning BY, Dass MS, Smalley MJ and
Pearson HB: The PI3K-AKT-mTOR pathway and prostate cancer: At the
crossroads of AR, MAPK, and WNT signaling. Int J Mol Sci.
21(4507)2020.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Bloedjes TA, de Wilde G, Maas C, Eldering
E, Bende RJ, van Noesel CJM, Pals ST, Spaargaren M and Guikema JEJ:
AKT signaling restrains tumor suppressive functions of FOXO
transcription factors and GSK3 kinase in multiple myeloma. Blood
Adv. 4:4151–4164. 2020.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Tsubaki M, Takeda T, Tomonari Y, Koumoto
YI, Imano M, Satou T and Nishida S: Overexpression of HIF-1α
contributes to melphalan resistance in multiple myeloma cells by
activation of ERK1/2, Akt, and NF-κB. Lab Invest. 99:72–84.
2019.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Wang L, Lin N and Li Y: The PI3K/AKT
signaling pathway regulates ABCG2 expression and confers resistance
to chemotherapy in human multiple myeloma. Oncol Rep. 41:1678–1690.
2019.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Schuster SR, Kortuem KM, Zhu YX, Braggio
E, Shi CX, Bruins LA, Schmidt JE, Ahmann G, Kumar S, Rajkumar SV,
et al: The clinical significance of cereblon expression in multiple
myeloma. Leuk Res. 38:23–28. 2014.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Kortüm KM, Mai EK, Hanafiah NH, Shi CX,
Zhu YX, Bruins L, Barrio S, Jedlowski P, Merz M, Xu J, et al:
Targeted sequencing of refractory myeloma reveals a high incidence
of mutations in CRBN and Ras pathway genes. Blood. 128:1226–1233.
2016.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Zhu YX, Braggio E, Shi CX, Kortuem KM,
Bruins LA, Schmidt JE, Chang XB, Langlais P, Luo M, Jedlowski P, et
al: Identification of cereblon-binding proteins and relationship
with response and survival after IMiDs in multiple myeloma. Blood.
124:536–545. 2014.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Bjorklund CC, Ma W, Wang ZQ, Davis RE,
Kuhn DJ, Kornblau SM, Wang M, Shah JJ and Orlowski RZ: Evidence of
a role for activation of Wnt/beta-catenin signaling in the
resistance of plasma cells to lenalidomide. J Biol Chem.
286:11009–11020. 2011.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Bjorklund CC, Baladandayuthapani V, Lin
HY, Jones RJ, Kuiatse I, Wang H, Yang J, Shah JJ, Thomas SK, Wang
M, et al: Evidence of a role for CD44 and cell adhesion in
mediating resistance to lenalidomide in multiple myeloma:
Therapeutic implications. Leukemia. 28:373–383. 2014.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Ocio EM, Fernández-Lázaro D, San-Segundo
L, López-Corral L, Corchete LA, Gutiérrez NC, Garayoa M, Paíno T,
García-Gómez A, Delgado M, et al: In vivo murine model of acquired
resistance in myeloma reveals differential mechanisms for
lenalidomide and pomalidomide in combination with dexamethasone.
Leukemia. 29:705–714. 2015.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Danziger SA, McConnell M, Gockley J, Young
MH, Rosenthal A, Schmitz F, Reiss DJ, Farmer P, Alapat DV, Singh A,
et al: Bone marrow microenvironments that contribute to patient
outcomes in newly diagnosed multiple myeloma: A cohort study of
patients in the total therapy clinical trials. PLoS Med.
17(e1003323)2020.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Yu G, Wang LG, Han Y and He QY:
clusterprofiler: An R package for comparing biological themes among
gene clusters. OMICS. 16:284–287. 2012.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Li H, Han D, Hou Y, Chen H and Chen Z:
Statistical inference methods for two crossing survival curves: A
comparison of methods. PLoS One. 10(e0116774)2015.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Mittal V: Epithelial mesenchymal
transition in tumor metastasis. Annu Rev Pathol. 13:395–412.
2018.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Uddin S, Bu R, Ahmed M, Abubaker J,
Al-Dayel F, Bavi P and Al-Kuraya KS: Overexpression of leptin
receptor predicts an unfavorable outcome in Middle Eastern ovarian
cancer. Mol Cancer. 8(74)2009.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Dimopoulos MA, Moreau P, Terpos E, Mateos
MV, Zweegman S, Cook G, Delforge M, Hájek R, Schjesvold F, Cavo M,
et al: Multiple myeloma: EHA-ESMO clinical practice guidelines for
diagnosis, treatment and follow-up†. Ann Oncol. 32:309–322.
2021.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Martinez-Hoyer S and Karsan A: Mechanisms
of lenalidomide sensitivity and resistance. Exp Hematol. 91:22–31.
2020.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Weber DM, Chen C, Niesvizky R, Wang M,
Belch A, Stadtmauer EA, Siegel D, Borrello I, Rajkumar SV,
Chanan-Khan AA, et al: Lenalidomide plus dexamethasone for relapsed
multiple myeloma in North America. N Engl J Med. 357:2133–2142.
2007.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Richardson P, Jagannath S, Hussein M,
Berenson J, Singhal S, Irwin D, Williams SF, Bensinger W, Badros
AZ, Vescio R, et al: Safety and efficacy of single-agent
lenalidomide in patients with relapsed and refractory multiple
myeloma. Blood. 114:772–778. 2009.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Strepkos D, Markouli M, Klonou A,
Papavassiliou AG and Piperi C: Histone Methyltransferase SETDB1: A
common denominator of tumorigenesis with therapeutic potential.
Cancer Res. 81:525–534. 2021.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Cao N, Yu Y, Zhu H, Chen M, Chen P, Zhuo
M, Mao Y, Li L, Zhao Q, Wu M and Ye M: SETDB1 promotes the
progression of colorectal cancer via epigenetically silencing p21
expression. Cell Death Dis. 11(351)2020.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Karanth AV, Maniswami RR, Prashanth S,
Govindaraj H, Padmavathy R, Jegatheesan SK, Mullangi R and
Rajagopal S: Emerging role of SETDB1 as a therapeutic target.
Expert Opin Ther Targets. 21:319–331. 2017.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Zhou Z, Wu B, Tang X, Yang W, Zou Q and
Wang H: High SET domain Bifurcated 1 (SETDB1) expression predicts
poor prognosis in breast carcinoma. Med Sci Monit.
26(e922982)2020.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Shang W, Wang Y, Liang X, Li T, Shao W,
Liu F, Cui X, Wang Y, Lv L, Chai L, et al: SETDB1 promotes gastric
carcinogenesis and metastasis via upregulation of CCND1 and MMP9
expression. J Pathol. 253:148–159. 2021.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Yu L, Ye F, Li YY, Zhan YZ, Liu Y, Yan HM,
Fang Y, Xie YW, Zhang FJ, Chen LH, et al: Histone methyltransferase
SETDB1 promotes colorectal cancer proliferation through the
STAT1-CCND1/CDK6 axis. Carcinogenesis. 41:678–688. 2020.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Guo C, Chu H, Gong Z, Zhang B, Li C, Chen
J and Huang L: HOXB13 promotes gastric cancer cell migration and
invasion via IGF-1R upregulation and subsequent activation of
PI3K/AKT/mTOR signaling pathway. Life Sci.
278(119522)2021.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Bakr AG, El-Bahrawy AH, Taha HH and Ali
FEM: Diosmin enhances the anti-angiogenic activity of sildenafil
and pentoxifylline against hepatopulmonary syndrome via regulation
of TNF-alpha/VEGF, IGF-1/PI3K/AKT, and FGF-1/ANG-2 signaling
pathways. Eur J Pharmacol. 873(173008)2020.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Chen C, Liu P, Duan X, Cheng M and Xu LX:
Deferoxamine-induced high expression of TfR1 and DMT1 enhanced iron
uptake in triple-negative breast cancer cells by activating
IL-6/PI3K/AKT pathway. Onco Targets Ther. 12:4359–4377.
2019.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Dou R, Qian J, Wu W, Zhang Y, Yuan Y, Guo
M, Wei R, Yang S, Jurczyszyn A, Janz S, et al: Suppression of
steroid 5alpha-reductase type I promotes cellular apoptosis and
autophagy via PI3K/Akt/mTOR pathway in multiple myeloma. Cell Death
Dis. 12(206)2021.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Xu Y, Feng X, Zhou Q, Jiang W, Dai Y,
Jiang Y, Liu X, Li S, Wang Y, Wang F, et al: Novel small molecular
compound AE-848 potently induces human multiple myeloma cell
apoptosis by modulating the NF-kappaB and PI3K/Akt/mTOR signaling
pathways. Onco Targets Ther. 13:13063–13075. 2020.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Ramakrishnan V and Kumar S: PI3K/AKT/mTOR
pathway in multiple myeloma: from basic biology to clinical
promise. Leuk Lymphoma. 59:2524–2534. 2018.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Li N, Liu B, Wang D, Song Y, Luo S and
Fang B: Preliminary study on the relationship among stem cell
markers, drug resistance and PI3K signaling pathway in multiple
myeloma (MM) cell. Transl Cancer Res. 9:3385–3391. 2020.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Zhang Y and Weinberg RA:
Epithelial-to-mesenchymal transition in cancer: Complexity and
opportunities. Front Med. 12:361–373. 2018.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Arumugam T, Ramachandran V, Fournier KF,
Wang H, Marquis L, Abbruzzese JL, Gallick GE, Logsdon CD, McConkey
DJ, Choi W, et al: Epithelial to mesenchymal transition contributes
to drug resistance in pancreatic cancer. Cancer Res. 69:5820–5828.
2009.PubMed/NCBI View Article : Google Scholar
|
|
53
|
McConkey DJ, Choi W, Marquis L, Martin F,
Williams MB, Shah J, Svatek R, Das A, Adam L, Kamat A, et al: Role
of epithelial-to-mesenchymal transition (EMT) in drug sensitivity
and metastasis in bladder cancer. Cancer Metastasis Rev.
28:335–344. 2009.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Huang J, Li H and Ren G:
Epithelial-mesenchymal transition and drug resistance in breast
cancer (Review). Int J Oncol. 47:840–848. 2015.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Ryu J, Koh Y, Park H, Kim DY, Kim DC, Byn
JM, Lee HJ and Yoon SS: Highly expressed integrin-α8 induces
epithelial to mesenchymal transition-like features in multiple
myeloma with early relapse. Mol Cells. 39:898–908. 2016.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Peng Y, Li F, Zhang P, Wang X, Shen Y,
Feng Y, Jia Y, Zhang R, Hu J and He A: IGF-1 promotes multiple
myeloma progression through PI3K/Akt-mediated
epithelial-mesenchymal transition. Life Sci.
249(117503)2020.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Hou Z, Sun L, Xu F, Hu F, Lan J, Song D,
Feng Y, Wang J, Luo X, Hu J and Wang G: Blocking histone
methyltransferase SETDB1 inhibits tumorigenesis and enhances
cetuximab sensitivity in colorectal cancer. Cancer Lett. 487:63–73.
2020.PubMed/NCBI View Article : Google Scholar
|