Introduction
Acromegaly is a rare disease, usually caused by a
pituitary tumor secreting growth hormone (GH), that has an annual
incidence rate between 0.2 and 1.1 cases per 100,000 individuals
(1). Given the clinical features
associated with this pathology develop insidiously, the delay
between onset and diagnosis can be >5 years (2). Numerous comorbidities are associated
with exposure to high levels of GH and its peripheral mediator,
insulin-like growth factor 1 (IGF-1).
Goiter is a frequent endocrine comorbidity of
acromegaly. It is the result of overstimulation by GH and IGF-1 of
the follicular epithelium (3,4).
Diffuse and nodular goiter represent ~57.7% of acromegaly cases,
while toxic nodular goiter is an unusual occurrence in acromegalic
patients, although it is more common in women (3,5).
Usually, patients with acromegaly are euthyroid or develop central
hypothyroidism due to the compressive nature of the tumor.
Hyperthyroidism is diagnosed in 3.5-26% of acromegalic patients
(3). Active acromegaly can impair
thyroid-stimulating hormone (TSH) response to thyrotropin-releasing
hormone (TRH), leading to abnormal TSH values, and increased
conversion of free thyroxine (T4) to free triiodothyronine (T3),
which results in central suppression of TSH secretion (6).
The association between acromegaly and thyroid
autoimmunity was also shown previously; 25% of the patients, the
majority of whom were women, had positive anti-thyroid peroxidase
(ATPO) antibodies (7).
Autoimmune thyroid diseases (AITD) affect 2-5% of
the population, being more commonly diagnosed in women, and these
include Graves' disease among other pathologies (8,9).
Clinical characteristics of Graves' disease are represented by
hyperthyroidism, diffuse goiter, and Graves' ophthalmopathy (GO)
(10). The primary clinical
presentation of GO includes eyelid retraction and periorbital edema
(11). Both signs may mimic soft
tissue proliferation and coarsening of the facial features,
characteristic of acromegaly (12). Other signs, such as increased
sweating, heat intolerance, visual disturbances, and fatigue, are
common both in hyperthyroidism and acromegaly (12). To summarize, a diagnosis of
acromegaly may be overlooked, due to certain shared symptoms with
Graves' disease.
Acromegaly and hyperthyroidism have distinct
pathophysiological mechanisms and hormonal profiles; biological
parameters, such as those related to phospho-calcium metabolism,
exhibit similar changes between the two diseases (12). In acromegaly, higher serum
phosphate (due to increased renal tubular resorption) and increased
plasma calcium levels, together with hypercalciuria, most likely as
a result of GH or IGF-I direct actions, are observed (12,13).
Hyperthyroidism is also associated with a catabolic state that
leads to bone resorption and hypercalcemia (14). High levels of serum calcium lower
the levels of parathormone and, as a consequence, increased renal
tubular reabsorption of phosphorus, leading to high levels of this
electrolyte are observed (14).
The clinical case presented here describes the
difficulties in diagnosing acromegaly due to its unusual
association with two distinct thyroid pathologies.
Case report
A 47-year-old female patient was admitted to the
Department of Endocrinology of the Emergency County Hospital
Timisoara in October 2018, with the following symptoms:
Hyperhidrosis, fatigue, paresthesia in the upper left limb,
dyspnea, dysphagia, and a gritty sensation in the eyes accompanied
by conjunctival edema and erythema.
The personal history of the patient revealed that
she was diagnosed with subclinical hyperthyroidism in 2012
(etiology not mentioned) and was treated with antithyroid drugs for
4 years. At that time, she was also diagnosed with hypertension and
received treatment with β-blockers and
angiotensin-converting-enzyme inhibitors. In 2016, the patient
underwent neck ultrasound and thyroid scintigraphy resulting in a
diagnosis of toxic adenoma and was thus treated with radioactive
iodine. Later that year she developed hypothyroidism, for which she
was given levothyroxine until September 2018. Therapy was
discontinued due to the lab results that revealed hyperthyroidism
and the patient was transferred to the development of bilateral
ophthalmopathy.
During the first admission to the Department of
Endocrinology (October 2018), the clinical examination of the
anterior cervical region revealed a palpable painless nodule of ~5
cm in the right lobe, which had increased consistency and was
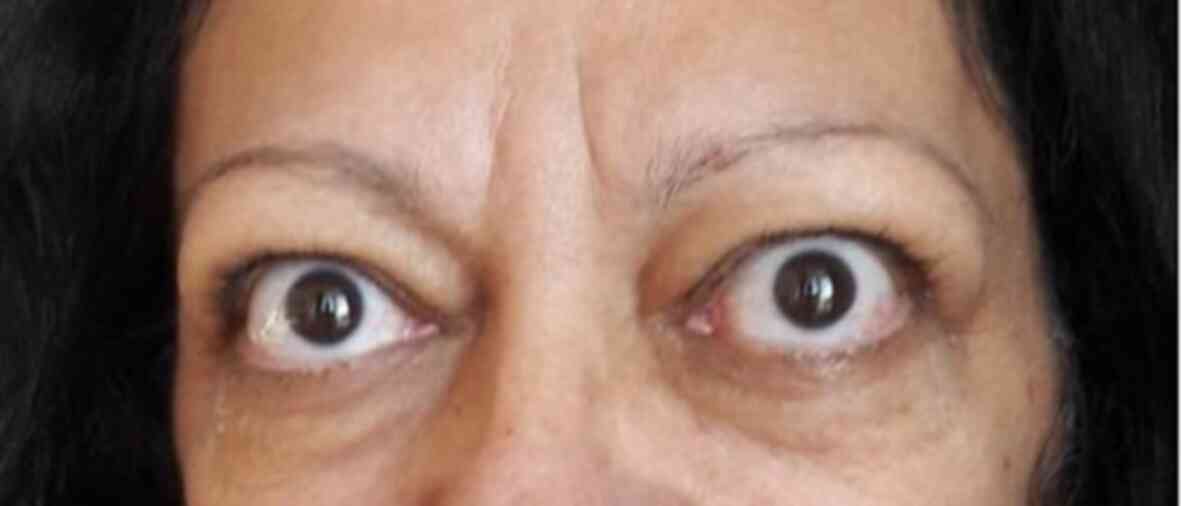
mobile while swallowing. The following abnormalities were also
noted: Bilateral symmetric exophthalmos and increased blood
pressure (160/80 mmHg).
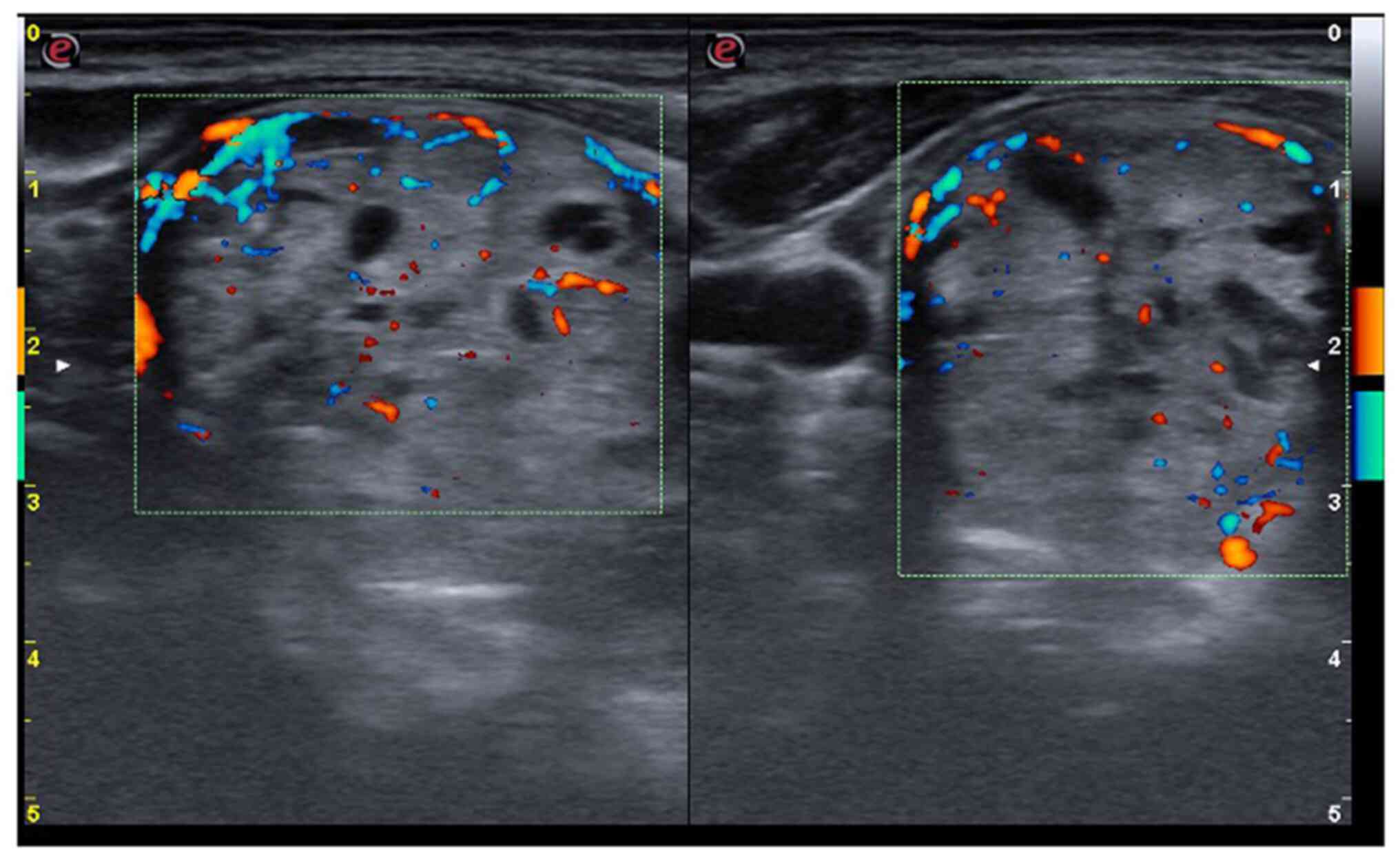
The neck ultrasound detected a hypoechoic thyroid
parenchyma with an increased volume of the thyroid (26.8 ml, upper
normal limit <16 ml). The right lobe was completely replaced by
a spongiform nodule, with increased vascularity (Fig. 1), which was classified as benign by
ACR-TIRADS (score 2) (15). The
left lobe exhibited increased vascularity and, close to the
posterior capsule, there was a partially cystic nodule of 6/5/7 mm
(Fig. 2), also classified as
benign by ACR-TIRADS (score 2).
Laboratory tests confirmed that the patient
presented subclinical hyperthyroidism, with low levels of TSH
(0.025 mU/l; normal range, 0.55-4.78 mU/l) and normal levels of FT4
(14.93 pmol/l; normal range, 11.50-22.70 pmol/l), and FT3 (5.98
pmol/l; normal range, 3.54-6.47 pmol/l). Given the medical history
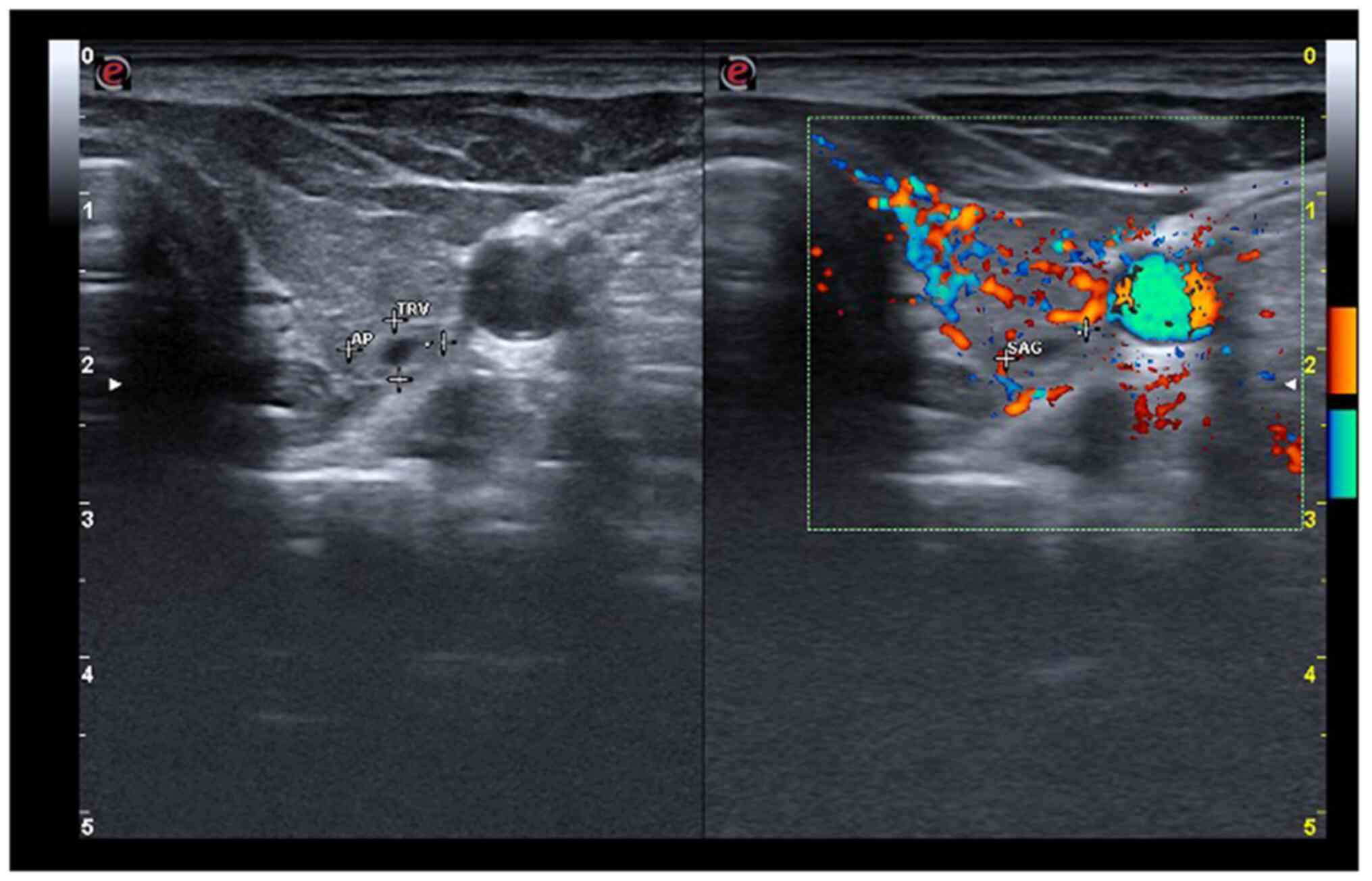
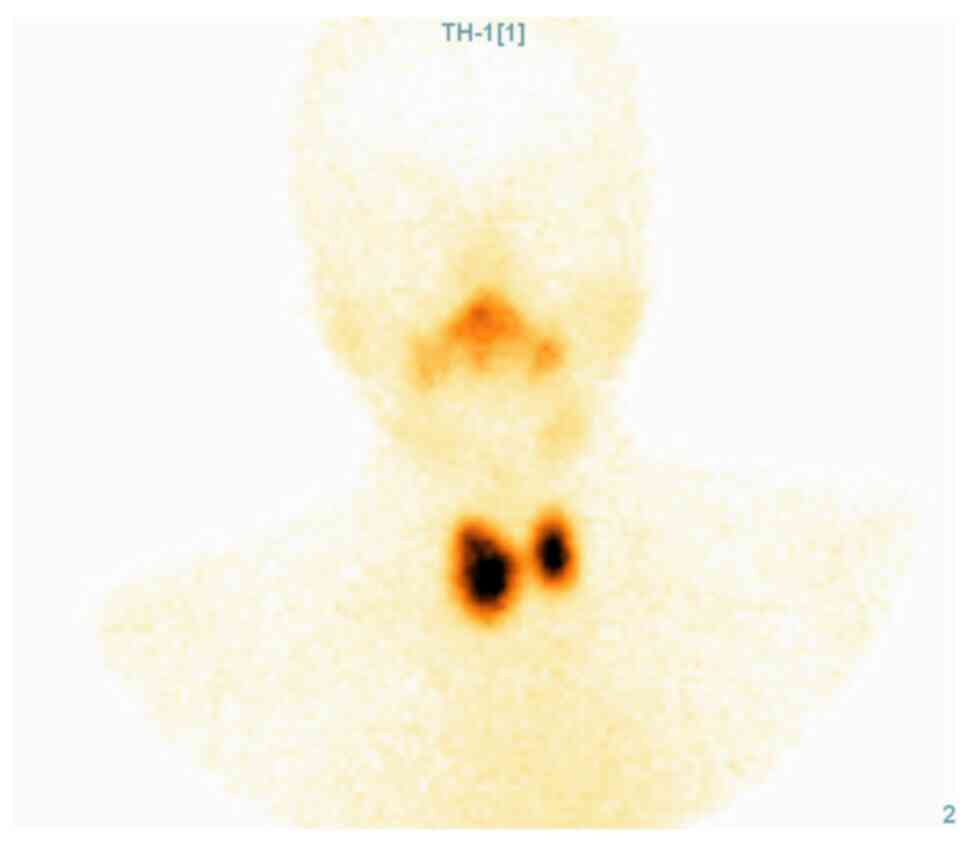
of the patient and the morphological aspect of the right lobe, a
thyroid scintigraphy was performed. It showed increased
Tc99m-pertechnetate uptake in the right lobe, relative
to the extra-nodular thyroid tissue, an aspect that confirmed the
diagnosis of toxic adenoma (Fig.
3).
The presence of the bilateral ophthalmopathy and
increased vascularity of the thyroid left lobe raised the suspicion
of Graves' disease, thus the levels of anti-TSH receptor antibodies
were measured; their high titer (>40 IU/l, normal values
<1.75 IU/l) confirmed this diagnosis. The patient had normal
values of ATPO antibodies (48 IU/ml; normal range, 0-60 IU/ml) and
high levels of antithyroglobulin antibodies (>500 IU/ml; normal
range, 0-60 IU/ml).
Other blood tests results were also abnormal:
Fasting blood glucose=124 mg/dl (normal range, 70-109 mg/dl),
glycated hemoglobin=6.1% (normal range, 4.36-5.6%), serum phosphate
5.8 and 6.1 mg/dl (tested twice; normal range, 2.5-4.9 mg/dl),
serum magnesium 1.48 mg/dl (normal range, 1.8-2.4 mg/dl) and
alkaline phosphatase 127 U/l (normal range, 46-116 U/l).
Given the fact that the patient denied the
consumption of drugs containing phosphate, the normal renal
function, and normal values of serum calcium, 25(OH)vitamin D, and
parathormone, the most common causes of increased serum phosphate
levels were excluded.
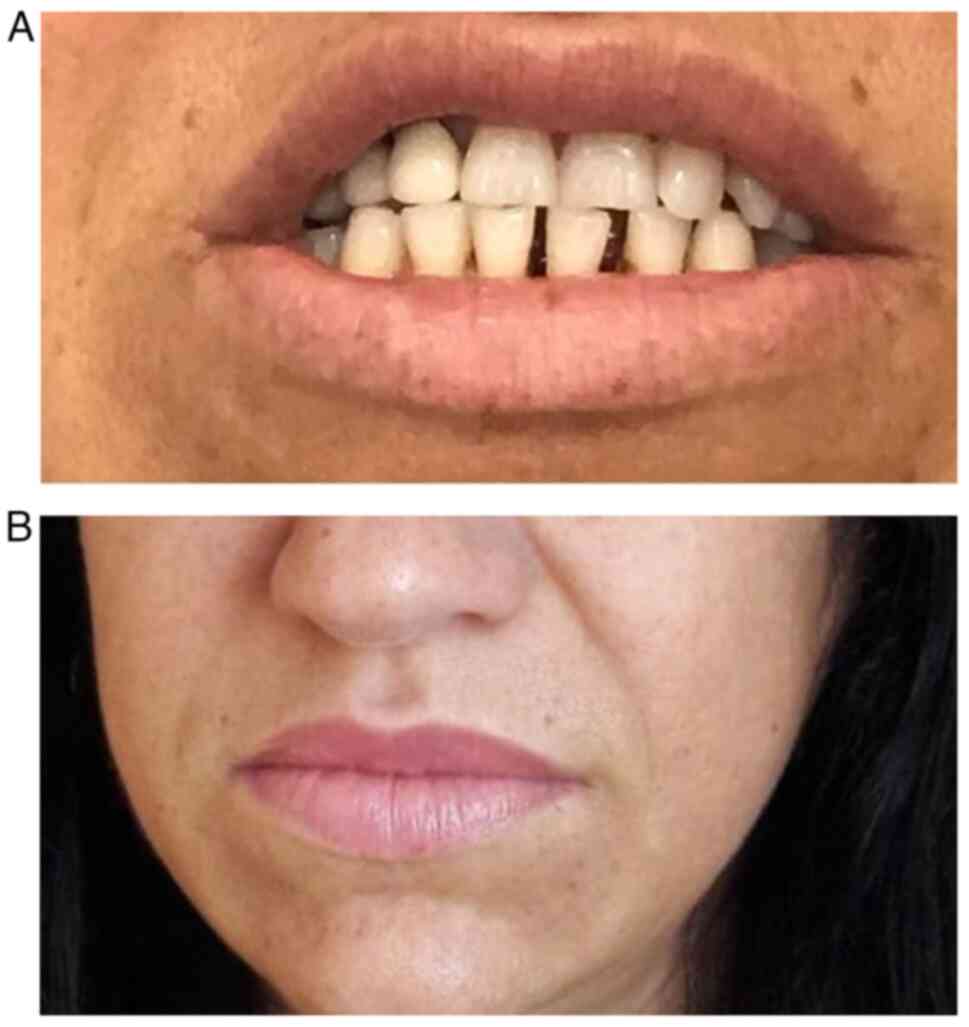
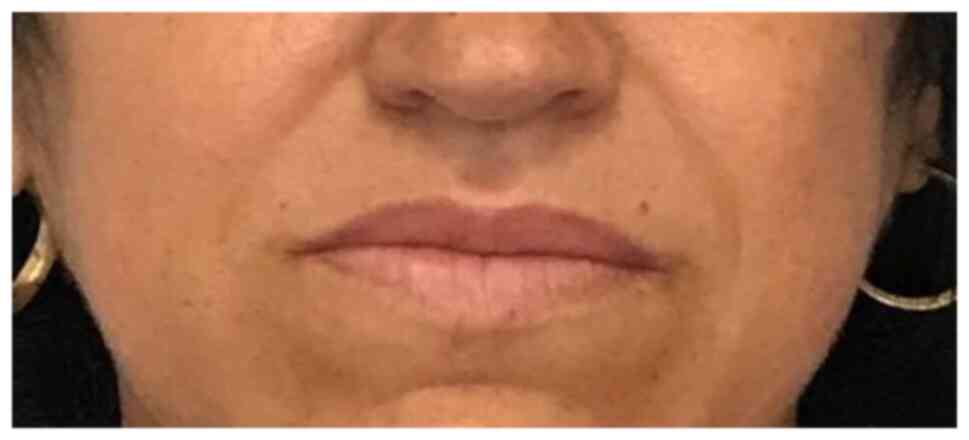
A more detailed history and targeted clinical exam
were performed. They revealed the following: Macroglossia,
increased interdental spaces (Fig.
4A), enlarged lips, and prominent nasolabial folds (Fig. 4B). The patient also mentioned she
was not able to take off her wedding ring for ~5 years. The
suspicion of acromegaly was raised and, consequently, IGF-1 (891
ng/ml, normal range for age and sex 83.3-220 ng/ml) and basal GH
(20.2 ng/ml, normal value for age and sex <10 ng/ml) were
determined. A suppression test with 75 g glucose administered
orally was also performed, showing the failure to suppress GH below
1 ng/ml and thus confirming the diagnosis of acromegaly. The
evaluation of the global function of the pituitary gland did not
identify other hormonal deficiencies (prolactin=4 ng/ml, normal
range=2.8-29.2 ng/ml; FSH=75.98 mIU/ml, normal range for
menopause=23.0-116.3 mIU/ml; LH=43.05 mIU/ml, normal range for
menopause=7.9-53.8 mIU/ml; estradiol <11.80 pg/ml, normal range
for menopause <32.2 pg/ml; and cortisol=12.38 mcg/dl, normal
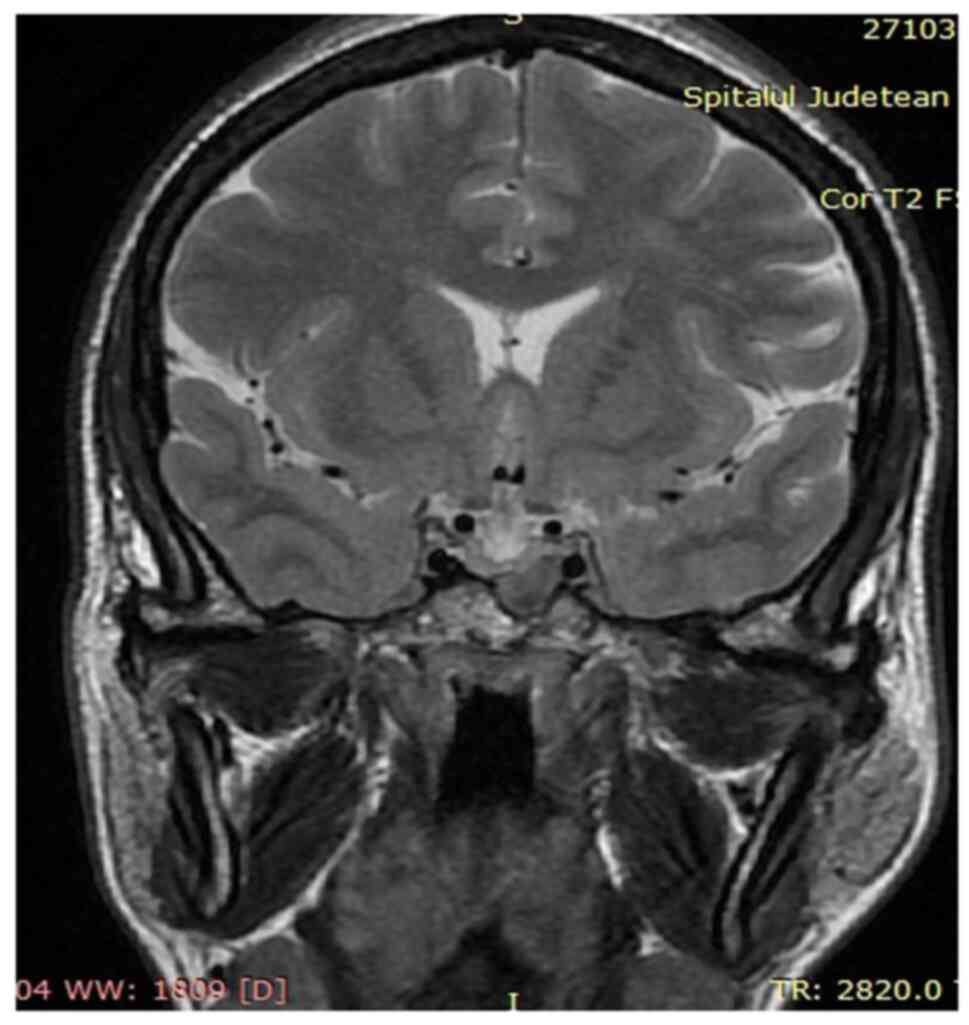
range=4.3-22.4 mcg/dl). A pituitary MRI was performed to detect and
characterize the adenoma. It revealed a macroadenoma (13/9 mm),
hypointense compared to the normal pituitary parenchyma (in
T2-weighted signal), which invaded the left sphenoid sinus, without
imprinting the optic chiasm (Fig.
5).
To treat the hyperthyroidism, the patient was
administered thiamazole 10 mg daily. Ophthalmopathy was classified
according to the European guidelines (16) as active (clinical activity score,
3) and mild (Fig. 6).
Consequently, a watchful strategy was implemented, and the patient
received local treatment. Therapy of the pituitary pathology was
prioritized and thyroid function and morphology were reassessed
thereafter. Surgery was recommended and the patient was referred to
the Neurosurgery Department.
The patient was readmitted to the Department of
Endocrinology in March 2019, 12 weeks after surgery
(transsphenoidal adenectomy). Lab tests revealed high levels of
IGF-1 (474 ng/ml, normal range 81.8-219 ng/ml), high levels of
basal GH (9.14 ng/ml, normal range 0.05-8 ng/ml), and lack of
suppression of GH <1 ng/ml during an oral glucose test with 75 g
glucose. MRI was performed and a residual tumor was identified in
the sphenoid sinus. The disease was considered active and therapy
with octreotide (a somatostatin analogue) 20 mg intramuscularly
(IM) every 28 days was initiated. The level of serum phosphate was
4.5 mg/dl (normal range, 2.5-4.9 mg/dl). Blood glucose and glycated
hemoglobin were normal, and the response to oral glucose (blood
glucose at 120 min=87 mg/dl) were normal, as well.
The patient was screened for complications
associated with acromegaly and was diagnosed with severe sleep
apnea. The other screening tests were unremarkable. The patient did
not present any gastrointestinal complications.
The re-evaluation of thyroid function revealed
hypothyroidism (low TSH levels associated with low FT4 levels),
thus thiamazole was discontinued (the last daily dose was 5 mg).
The thyroid aspects in ultrasound and the status of the
ophthalmopathy did not change.
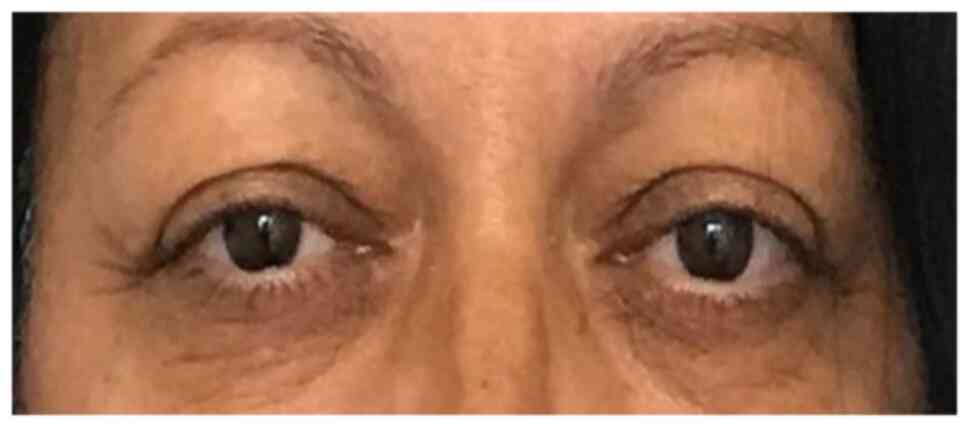
In June 2019, 3 months after therapy with
somatostatin analogues had been started, the patient was
re-evaluated. Even though the clinical status improved (Fig. 7), the response to therapy was poor.
Lab tests revealed high levels of IGF-1 (658 ng/ml, normal range
81.8-219 ng/ml), high levels of GH (8.49 ng/ml, normal range 0.05-8
ng/ml), and a lack of suppression of GH below 1 ng/ml during an
oral glucose tolerance test; MRI remained unchanged. Taking into
consideration the blood tests and the presence of the residual
tumor, the dose of octreotide was increased (30 mg IM every 28
days) and cabergoline was added (2 mg orally every week).
The function and morphology of the thyroid were
reassessed, showing mild subclinical hyperthyroidism, while the
neck ultrasound was unchanged. The ophthalmological consultation
classified the eye disease as inactive and moderate (clinical score
activity=1/10, Fig. 8) (16). Due to the history the thyroid
disease and the tracheal compression by the thyroid nodule (proven
by radiological studies), the patient was referred to an endocrine
surgeon for total thyroidectomy. After surgery, the evolution was
favorable and therapy with levothyroxine was initiated. The
pathological result confirmed the concurrence of two distinct
thyroid diseases in addition to the degenerative changes induced by
therapy, a hyperfunctioning goiter with pronounced degenerative
changes, imprecisely contoured adenomatous nodules, with areas of
hyalinization and interstitial calcification, cystic degeneration,
and areas of angiomatosis primarily expressed in the right lobe of
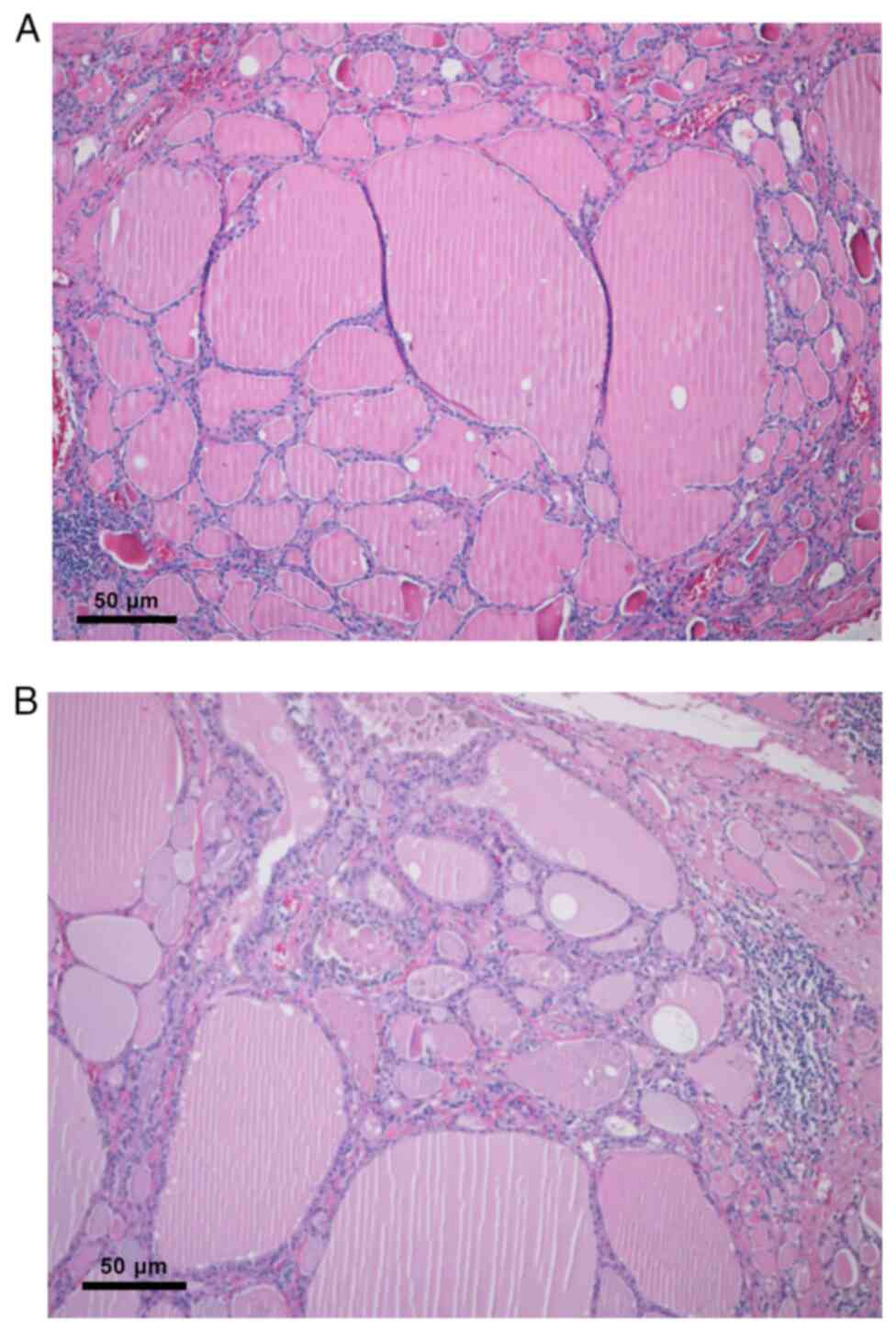
the thyroid (Fig. 9A and B).
The patient was re-evaluated periodically and, 9
months later, she was unresponsive to medical treatment. At this
point, pegvisomant (up to 20 mg daily), was initiated which was
well tolerated. After 6 months of therapy, IGF-1 was normal, and
the clinical status of the patient improved significantly. The
patient did not exhibit any adverse reactions, and the pituitary
tumoral rest did not increase.
Discussion
Here, the case of an acromegalic patient, who was
initially admitted to our department for hyperthyroidism, is
described. After routine lab tests, acromegaly was suspected and
confirmed thereafter by a dedicated workup. The etiology of
hyperthyroidism was challenging due to the coexistence of both
toxic adenoma and complicated Graves' disease. Secondary
hypothyroidism was excluded due to the medical history of the
patient, normal FT4 levels, positive anti-TSH receptor antibodies,
thyroid ultrasound (thyroid volume was increased), and scintigraphy
showing increased uptake in the right lobe (17,18).
As a result of the insidious evolution of
acromegaly, its signs and symptoms may remain unnoticed for long
periods of time. In our case, due to the concomitant thyroid
pathology, certain aspects of the clinical exam that suggested a
diagnosis of acromegaly in a patient who had no complaints related
to tumoral compression (headache, blurred vision, or vomiting) were
overlooked. During routine lab workouts, a high level of serum
phosphorus, accompanied by normal calcium and parathormone values,
leads to a more detailed anamnesis and physical exam. After asking
questions that targeted the signs and symptoms of acromegaly, the
diagnosis was considered more seriously. The association between
acromegaly and Graves' disease is rare, but may be due to a series
of clinical manifestations such as acral enlargement, soft tissue
overgrowth, coarsening of facial features, and joint pain. If these
manifestations are discrete or absent, as was described in the
present case, the diagnosis can be challenging. In such situations,
even a minimal biological change, such as hyperphosphatemia, should
receive more attention.
Hyperphosphatemia is not uncommon among these
patients, due to the increased tubular reabsorption of this
electrolyte, given the action of IGF-1 on the apical membrane
sodium-phosphate IIa cotransporters (19). It was hypothesized that phosphate
may reflect the status of acromegaly, but in patients with
preoperative serum phosphate >4.5 mg/dl, no correlation was
found (20). Nevertheless, its
decrease after 3 months may predict remission in patients with
discordant GH and IGF-1 values (20). Conversely, a study published in
2017 revealed that hyperphosphatemia at diagnosis was more common
in patients who had an active disease after therapy (21).
Acromegaly is a rare disease; however, its
association with thyroid pathology is not uncommon (3). Nevertheless, the coexistence of
Graves' disease and acromegaly has a very low prevalence (<1%)
(22). IGF-1 has a synergic effect
with TSH on thyroid cell proliferation and may mediate an
immunological response by stimulating the proliferation of T cells
and B cells, while GH might stimulate the production of TSH
receptor antibodies (22). All
these mechanisms play a role in thyroid growth and can influence
the severity of Graves' disease.
The presence of both toxic adenoma and Graves'
disease (Marine-Lenhart syndrome) is a very rare occurrence, with a
prevalence of 1-2.5% (23), and
their association with acromegaly has not previously been reported
in the literature, to the best of our knowledge. Given the fact
that the patient did not undergo a full medical check-up prior to
being referred to our department, it was suggested that the
radioiodine therapy may have induced Graves' disease (24). A study that included 191 patients
with nontoxic goiter treated with 131I found that 5% of
the patients developed hyperthyroidism without eye involvement
after 3 months of therapy, and this was more frequent in those who
had elevated ATPO antibody levels. The suggested mechanism that
induced autoimmunity was the release of antigens from the
follicular cells (25). A
retrospective study in patients treated with 131I for
toxic nodular goiter reported a prevalence of hyperthyroidism
associated with high levels of anti-TSH receptor antibodies in 4%
of the cases (26). Several risk
factors are associated with hyperthyroidism, such as genetic
predisposition and high levels of ATPO antibodies before therapy
(25). In our patient, titers of
ATPO antibodies were not evaluated before 131I therapy,
but they were increased when they were measured during the first
hospital admission. In addition, the patient had high levels of
antithyroglobulin antibodies, thus this type of antibody as a risk
factor for developing an autoimmune disease in this case could not
be excluded.
Orbitopathy is a known extrathyroidal complication
of Graves' disease, with an estimated incidence of 16 cases in
100,000 women, usually being present at the onset of the disease
(27). The patient in the present
case report provided little to no information regarding the onset
of her eye symptoms, thus we were not able to establish a timeline
regarding its evolution. A study by Coutu et al (28) found that high levels of GH were
associated with extraocular muscle enlargement, a situation that
may have been present in our case. After pituitary surgery, the
condition of our patient's eye disease improved, without specific
medication, an aspect also mentioned in the aforementioned
study.
Hypertension is a common complication of acromegaly,
with a prevalence of 50% in patients with active disease (29). Its severity is related to the
excess of GH and IGF-1, which cause expansion of plasma volume and
increased systemic vascular resistance, leading to elevated
diastolic blood pressure (29).
Conversely, hypertension caused by hyperthyroidism is mostly due to
increased systolic blood pressure as a result of renin release and
sodium reabsorption (30). The
patient in the present report had high blood pressure, most likely
due to intricate mechanisms related to acromegaly and
hyperthyroidism (stimulation of smooth muscle cell growth,
increased arterial stiffness, renin release and sodium
reabsorption) (29,30). Her blood pressure lowered, and the
dosage of the oral medication was reduced after treating the
acromegaly and hyperthyroidism.
Diagnosing and treating the patient, in this case,
was a challenge. She did not respond to somatostatin analogues,
even though she had predictors for a better evolution, such as
being a female of childbearing potential and presenting a
hypointense T2-weighted MRI signal of pituitary adenoma (31,32).
There are several factors that can influence the response to
somatostatin analogues, such as a lack of somatostatin receptors
type 2 and 5, Ki-67 nuclear protein, or cytokeratin staining
pattern (33). However, they could
not be analyzed in the present case.
It has been noted that 25% of the patients do not
respond to somatostatin analogues after 12 months (31). According to the current guidelines,
our patient was completely unresponsive to therapy (34). Given the fact that her glucose
metabolism was impaired before surgery and that she did not present
any criterion for remission after the therapy with somatostatin
analogues, we chose to initiate pegvisomant. ACROSTUDY showed that
a dose of 18 mg/day was effective in 65% of patients treated with a
GH receptor antagonist (35). In
addition, complications associated with the treatment were very
rare: 2.2% of patients reported local discomfort, lipoatrophy, or
reversible lipohypertrophy; 3-5% had tumoral growth; and 9%
presented liver enzyme abnormalities (34,35).
In the present case, the patient responded well to the therapy and
did not show any side effects.
Due to the presence of multiple endocrine
pathologies associated with glandular hyperfunction, the presence
of a genetic defect in G-protein-coupled receptors cannot be
excluded. However, its more frequent association with congenital
endocrine pathologies, as well as the absence of a particular
phenotype of the patient, made this hypothesis unlikely (36).
In conclusion, acromegaly remains difficult to
diagnose and treat, because it has a slow evolution and some of the
clinical findings might be assigned to the patient's comorbidities.
In such cases, all the clinical and biochemical abnormalities that
may suggest a diagnosis of acromegaly should be considered.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
MV, ISP, FV, MB, IG, AV, DA and MC collected,
analyzed and interpreted the data. ISP, MB, IG, DA, AV, and MV
wrote and critically revised the manuscript. All authors have read
and approved the final manuscript. All authors agreed to be
accountable for all aspects of the work. ISP, MV, IG, and AV
confirm the authenticity of all the raw data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
The patient provided written consent for the
publication of their data.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Crisafulli S, Luxi N, Sultana J, Fontana
A, Spagnolo F, Giuffrida G, Ferraù F, Gianfrilli D, Cozzolino A,
Cristina De Martino M, et al: Global epidemiology of acromegaly: A
systematic review and meta-analysis. Eur J Endocrinol. 185:251–263.
2021.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Esposito D, Ragnarsson O, Johannsson G and
Olsson DS: Prolonged diagnostic delay in acromegaly is associated
with increased morbidity and mortality. Eur J Endocrinol.
182:523–531. 2020.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Dąbrowska AM, Tarach JS, Kurowska M and
Nowakowski A: Thyroid diseases in patients with acromegaly. Arch
Med Sci. 10:837–845. 2014.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Vargas-Ortega G, Romero-Gameros CA,
Rendón-Macias ME, Balcázar-Hernández L, Sosa-Eroza E, Mercado M, de
Los Monteros-Sánchez ALE, Pérez-Aguilar B, Paredes-Manjarrez C,
Reyes-Olhagaray FB, et al: Risk factors associated with thyroid
nodular disease in acromegalic patients: A case-cohort study in a
tertiary center. Growth Horm IGF Res. 60-61(101431)2021.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Natchev E, Vandeva S, Kovatcheva R,
Kirilov G, Kalinov K and Zacharieva S: Thyroid gland changes in
patients with acromegaly. Arch Endocrinol Metab. 64:269–275.
2020.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Tirosh A and Shimon I: Complications of
acromegaly: Thyroid and colon. Pituitary. 20:70–75. 2017.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Manavela M, Vigovich C, Danilowicz K, Juri
A, Miechi L, Fernandez Valoni V and Bruno OD: Thyroid autoimmune
disorders in patients with acromegaly. Pituitary. 18:912–915.
2015.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Franco JS, Amaya-Amaya J and Anaya JM:
Thyroid disease and autoimmune diseases. In: Autoimmunity: from
bench to bedside. Anaya JM, Shoenfeld Y, Rojas-Villarraga A, et
al (eds). El Rosario University Press, Bogota, pp537-562,
2013.
|
|
9
|
Stanciu M, Bera LG, Popescu M, Grosu F and
Popa FL: Hashimoto's thyroiditis associated with thyroid adenoma
with Hürthle cells-case report. Rom J Morphol Embryol. 58:241–248.
2017.PubMed/NCBI
|
|
10
|
Pokhrel B and Bhusal K: Graves' Disease.
In: StatPearls [Internet]. StatPearls Publishing, Treasure Island,
FL, pp4-5, 2022.
|
|
11
|
Bahn RS: Graves' ophthalmopathy. N Engl J
Med. 362:726–738. 2010.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Gardner DG and Shoback D: Greenspan's
Basic and Clinical Endocrinology. 10th edition. McGraw Hill
Medical, New York, NY, pp69-120, 2017.
|
|
13
|
Halse J and Haugen HN: Calcium and
phosphate metabolism in acromegaly. Acta Endocrinol (Copenh).
94:459–467. 1980.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Mosekilde L and Christensen MS: Decreased
parathyroid function in hyperthyroidism: Interrelationships between
serum parathyroid hormone, calcium-phosphorus metabolism and
thyroid function. Acta Endocrinol (Copenh). 84:566–575.
1977.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Tessler FN, Middleton WD, Grant EG, Hoang
JK, Berland LL, Teefey SA, Cronan JJ, Beland MD, Desser TS, Frates
MC, et al: ACR thyroid imaging, reporting and data system
(TI-RADS): White paper of the ACR TI-RADS Committee. J Am Coll
Radiol. 14:587–595. 2017.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Bartalena L, Kahaly GJ, Baldeschi L, Dayan
CM, Eckstein A, Marcocci C, Marinò M, Vaidya B and Wiersinga WM:
EUGOGO†. The 2021 European Group on Graves' orbitopathy (EUGOGO)
clinical practice guidelines for the medical management of Graves'
orbitopathy. Eur J Endocrinol. 185:G43–G67. 2021.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Bel Lassen P, Kyrilli A, Lytrivi M and
Corvilain B: Graves' disease, multinodular goiter and subclinical
hyperthyroidism. Ann Endocrinol (Paris). 80:240–249.
2019.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Persani L: Clinical review: Central
hypothyroidism: Pathogenic, diagnostic, and therapeutic challenges.
J Clin Endocrinol Metab. 97:3068–3078. 2012.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Jehle AW, Forgo J, Biber J, Lederer E,
Krapf R and Murer H: IGF-I and vanadate stimulate Na/Pi-cotransport
in OK cells by increasing type II Na/Pi-cotransporter protein
stability. Pflugers Arch. 437:149–154. 1998.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Xie T, Tian P, Wu S, Zhang X, Liu T, Gu Y,
Sun C and Hu F: Serum phosphate: Does it more closely reflect the
true state of acromegaly? J Clin Neurosci. 71:26–31.
2020.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Yalin GY, Tanrikulu S, Gul N, Uzum AK,
Aral F and Tanakol R: Utility of baseline serum phosphorus levels
for predicting remission in acromegaly patients. J Endocrinol
Invest. 40:867–874. 2017.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Di Cerbo A, Pezzuto F and Di Cerbo A:
Growth hormone and insulin-like growth factor 1 affect the severity
of Graves' disease. Endocrinol Diabetes Metab Case Rep.
2017:17–0061. 2017.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Ribeiro F, Ruas L, Júnior A, Sousa A,
Araújo A, Mwambire J, Jesus A, Bussuan R, Sá L and Arbex A: Graves'
disease and Marine Lenhart syndrome: A rare clinical presentation.
Health. 11:1169–1176. 2019.
|
|
24
|
Shen G, Cui F, Huang R and Kuang A:
Graves' disease following radioiodine therapy for toxic adenoma:
Clinical case report. Medicine (Baltimore).
96(e8550)2017.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Nygaard B, Knudsen JH, Hegedüs L, Scient
AV and Hansen JE: Thyrotropin receptor antibodies and Graves'
disease, a side-effect of 131I treatment in patients with nontoxic
goiter. J Clin Endocrinol Metab. 82:2926–2930. 1997.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Nygaard B, Faber J, Veje A, Hegedüs L and
Hansen JM: Transition of nodular toxic goiter to autoimmune
hyperthyroidism triggered by 131I therapy. Thyroid. 9:477–481.
1999.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Lazarus JH: Epidemiology of Graves'
orbitopathy (GO) and relationship with thyroid disease. Best Pract
Res Clin Endocrinol Metab. 26:273–279. 2012.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Coutu B, Alvarez DA, Ciurej A, Moneymaker
K, White M, Zhang C and Drincic A: Extraocular muscle enlargement
in growth hormone-secreting pituitary adenomas. AJNR Am J
Neuroradiol. 43:597–602. 2022.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Ramos-Leví AM and Marazuela M: Bringing
cardiovascular comorbidities in acromegaly to an update. How should
we diagnose and manage them? Front Endocrinol (Lausanne).
10(120)2019.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Prisant LM, Gujral JS and Mulloy AL:
Hyperthyroidism: A secondary cause of isolated systolic
hypertension. J Clin Hypertens (Greenwich). 8:596–599.
2006.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Colao A, Auriemma RS, Lombardi G and
Pivonello R: Resistance to somatostatin analogs in acromegaly.
Endocr Rev. 32:247–271. 2011.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Puig-Domingo M, Resmini E, Gomez-Anson B,
Nicolau J, Mora M, Palomera E, Martí C, Halperin I and Webb SM:
Magnetic resonance imaging as a predictor of response to
somatostatin analogs in acromegaly after surgical failure. J Clin
Endocrinol Metab. 95:4973–4978. 2010.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Ezzat S, Caspar-Bell GM, Chik CL, Denis
MC, Domingue MÈ, Imran SA, Johnson MD, Lochnan HA, Grégoire Nyomba
BL, Prebtani A, et al: Predictive markers for postsurgical medical
management of acromegaly: A systematic review and consensus
treatment guideline. Endocr Pract. 25:379–393. 2019.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Katznelson L, Laws ER Jr, Melmed S,
Molitch ME, Murad MH, Utz A and Wass JA: Endocrine Society.
Acromegaly: An endocrine society clinical practice guideline. J
Clin Endocrinol Metab. 99:3933–3951. 2014.PubMed/NCBI View Article : Google Scholar
|
|
35
|
van der Lely AJ, Biller BM, Brue T,
Buchfelder M, Ghigo E, Gomez R, Hey-Hadavi J, Lundgren F, Rajicic
N, Strasburger CJ, et al: Long-term safety of pegvisomant in
patients with acromegaly: Comprehensive review of 1288 subjects in
ACROSTUDY. J Clin Endocrinol Metab. 97:1589–1597. 2012.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Fukami M, Suzuki E, Igarashi M, Miyado M
and Ogata T: Gain-of-function mutations in G-protein-coupled
receptor genes associated with human endocrine disorders. Clin
Endocrinol. 88:351–359. 2018.PubMed/NCBI View Article : Google Scholar
|