Introduction
Renal interstitial fibrosis, a progressive
pathological phenomenon, is a prominent feature of chronic kidney
disease in humans and animal models (1,2).
Renal interstitial fibrosis is also the final common pathway
leading to end-stage kidney disease (3). Chronic renal inflammation promotes
the progression of renal fibrosis by stimulating leukocyte
infiltration into the kidneys and activating intrinsic renal cells
to release profibrotic factors that drive renal fibrosis (4). Therefore, regulating inflammation is
a key factor in treating renal fibrosis (5).
NOD-like receptor thermal protein domain associated
protein 3 (NLRP3) forms a multi-protein immune complex called the
NLRP3 inflammasome, which is composed of NLRP3,
apoptosis-associated speck-like protein containing a caspase
recruitment domain (ASC) and pro-caspase-1(6). NLRP3 inflammasome assembly is
triggered by multiple signaling pathways, including those related
to lysosomal disruption, reactive oxygen species (ROS) and
extracellular ATP (7). NLRP3
inflammasome assembly activates the cysteine protease caspase-1,
which results in the secretion of mature, biologically active
IL-1β, thereby playing a central role in inflammation (8). NLRP3 inflammasome activation can
occur in both immune and resident cells of the glomerulus. Previous
studies using diverse animal models of kidney disease have shown
that the NLRP3 inflammasome exerts important effects on renal
inflammation and fibrosis (9-11).
Kim et al (12) have
demonstrated, using the unilateral ureteral obstruction (UUO)
model, that NLRP3-knockout mice exhibit reduced renal fibrosis and
inflammation compared with wild-type mice. Lysosomal cathepsins are
mainly located in lysosomes but can also function in the cell
plasma or extracellular matrix. To date, 15 human lysosomal
cathepsins have been reported, of which cathepsin B, D, S and L
play an important role in renal physiopathology. Furthermore, NLRP3
inflammasome activation promotes renal inflammation, leading to
glomerular damage and end-stage kidney disease in chronic kidney
disease models (13-15).
The expression of lysosomal cathepsins is significantly elevated in
UUO mice, suggesting that pharmacological inhibition of cathepsins
may lead to the alleviation of kidney fibrosis (14). In addition, small interfering
(si)RNA knockdown of cathepsins B, L and S (CTSB, CTSL and CTSS,
respectively) inhibits NLRP3 inflammasome activation by
downregulating the production of pro-caspase-1 and pro-IL-1β in
HK-2 cells (human tubular epithelial cell line) and mouse knockout
models (16,17). Therefore, cathepsin-mediated NLRP3
inflammasome activation may be a therapeutic target in renal
fibrosis.
Fluorofenidone
[1-(3-fluorophenyl)-5-methyl-2-(1H)-pyridone; AKFPD] is
a novel low-molecular-weight pyridone agent. AKFPD exerts
anti-inflammatory effects by reducing oxidative stress and
apoptosis, ultimately suppressing fibrosis in the kidneys, liver
and lungs (18-22).
Fluorofenidone is associated with decreased inflammatory burden
(23). By contrast, conditions
that cause renal fibrosis, diabetic nephropathy for example, are
associated with high burden of inflammation (24). A number of inflammatory markers,
including C-reactive protein (25), serum uric acid (26) and systemic inflammatory index
(27), are increased in diabetic
nephropathy. Hence, studying the effects of Fluorofenidone in renal
fibrosis makes sense. Our previous study demonstrated that AKFPD
reduces IL-1β production by suppressing NLRP3 inflammasome
activation in UUO rats, suggesting that AKFPD is a new
anti-inflammatory agent (28).
Furthermore, a recent study has reported that AKFPD attenuates
renal fibrosis by inhibiting the mitochondrial ROS (mtROS)-NLRP3
pathway in a murine model of folic acid nephropathy (29). Nonetheless, the effect of AKFPD on
the NLRP3 inflammasome in renal intrinsic cells remains unclear. In
addition, the potential underlying mechanisms of the suppression of
NLRP3 inflammasome activation by AKFPD in renal inflammation remain
largely unknown. Therefore, the present study aimed to investigate
the underlying mechanism of AKFPD in suppressing NLRP3 inflammasome
activation in UUO rats. The study findings provided insights into
understanding the anti-inflammatory and anti-fibrotic effects of
AKFPD against renal fibrosis.
Materials and methods
Ethics approval
The present study protocol was reviewed and approved
by the Medical Research Ethics Committee of The First Affiliated
Hospital, Jiangxi Medical College, Nanchang University (Nanchang,
China; approval no. 2020-1-59).
Animals and experimental protocol
Male Sprague Dawley rats (8 weeks old; 180-220 g;
n=30) were purchased from Silaike Laboratory Animal Co. Ltd. The
rats were housed in a pathogen-free environment at 25±2˚C with
55±2% humidity and under a 12-h light/dark cycle and provided a
standard diet and tap water ad libitum. The rats were
randomly divided into six groups: The 3- and 7-day sham-operated
(sham group), 3- and 7-day UUO model (UUO group), and 3- and 7-day
AKFPD treatment groups (AKFPD group), with five rats in each group.
The UUO model was established by ligating the left ureter in the
UUO and AKFPD groups, as previously described (30). Similar surgical procedures were
performed in the sham group; however, the left ureter was only
exposed and not ligated. AKFPD (lot no. 20190810) was obtained from
the Haikou Pharmaceutical Factory Co., Ltd. The rats in the sham
and UUO groups were gavaged daily with 0.5% sodium carboxymethyl
cellulose (cat. no. 11926043; Shanghai Aladdin Biochemical
Technology Co., Ltd.), whereas the rats in the AKFPD group received
500 mg/kg/day AKFPD dissolved in 0.5% sodium carboxymethyl
cellulose. AKFPD and the vehicle were administered starting day 1
after surgery until the day of euthanasia. The rats were
administered 1% sodium pentobarbital (cat. no. 0123A001;
MilliporeSigma) and sacrificed 3 and 7 days after surgery. Rats
were anesthetized by intraperitoneal injection of sodium
pentobarbital (60 mg/kg) for UUO operation. Rats were sacrificed by
intraperitoneal injection of overdoses of sodium pentobarbital (150
mg/kg) for harvesting renal tissue. A portion of the left kidney
was excised, and samples were fixed in 10% neutral-buffered
formalin and 2.5% glutaraldehyde phosphate buffer (pH 7.4) for
pathological examination. The remaining kidney tissue was preserved
in liquid nitrogen for further experiments.
Histopathology
The kidney tissue samples were fixed with 4%
paraformaldehyde for 24 h at 4˚C. A section of the harvested left
kidney was soaked in 70 and 80% alcohol for 30 min each, followed
by soaking in 95% ethanol for 30 min (x2 times) and anhydrous
ethanol for 30 min, and then soaked in xylene for 15 min. Finally,
the sample was placed in the embedding box and soaked in wax at
60˚C for 2 h. The paraffin-embedded kidney tissue was sliced into 4
µm-thick sections and stained with hematoxylin-eosin (H&E) and
Masson's trichrome. For H&E staining, a tissue section was
incubated with hematoxylin for 10 min and eosin for 5 min, both at
room temperature, and rinsed under running water. Masson trichrome
staining was performed using the Trichrome Stain (Masson) Kit (cat.
no. G1006; Wuhan Servicebio Technology Co., Ltd.) at room
temperature, apart from Masson A staining, which was incubated for
30 min at 65˚C. Tubulointerstitial injury was quantified based on
the degree of renal interstitial fibrosis, tubular dilatation,
atrophy, tubular epithelial injury, inflammatory cell infiltration
and interstitial edema. Tubulointerstitial fibrosis was assessed in
Masson's trichrome-stained sections. H&E staining sections were
randomly selected from 5 renal interstitial fields, including upper
left, lower left, middle, upper right and lower right, under an
optical microscope at a x200 magnification. The renal interstitial
injury score was assigned in a single blinded manner. The H&E
score and tubulointerstitial fibrosis degree were determined as
previously described (31). The
samples were visualized using an Olympus light microscope (BX43;
Olympus Corporation).
Enzyme-linked immunosorbent assay
(ELISA)
Levels of TNF-α and IL-1β in kidney homogenates were
determined using specific ELISA kits (cat. nos. MM-0180R1,
MM-0047R1; Elabscience Biotechnology, Inc.) according to the
manufacturer's instructions.
HK-2 cell culture and treatment
HK-2 cell line was purchased from Beijing Beina
Chuanglian Biotech Institute (cat. no. BNCC295660). The cells were
cultured in a mixture of Dulbecco's modified Eagle medium and Ham's
F-12 medium (Nanjing KeyGen Biotech Co., Ltd.) supplemented with
10% fetal bovine serum (FBS), 100 U/ml penicillin, and 100 µg/ml
streptomycin under a humidified atmosphere of 5% CO2 and
95% O2 at 37˚C. The cells were seeded in six-well
culture plates and cultured in a complete medium. The plates were
randomly divided into three groups: Normal (N),
hypoxia/reoxygenation (H/R) and AKFPD. The cells in the AKFPD and
H/R groups were cultured for 12 h at 37˚C in a medium without
nutrients (serum- and glucose-free) under hypoxic conditions (1%
O2, 94% N2 and 5% CO2) to induce
hypoxic injury, and the cells were then transferred back to a
regular culture medium with oxygen for 6 h at 37˚C to facilitate
reoxygenation. The normal group cells were incubated in a complete
culture medium under regular incubator conditions (5%
CO2 and 95% air). The cells in the AKFPD group were
pre-incubated at 37˚C with 400 µg/ml AKFPD for 24 h.
Isolation and culture of
peritoneal-derived macrophages (PDMs)
PDMs were isolated from 50 male C57BL/6 mice (6-8
weeks old, 20-25 g), purchased from Shanghai SLAC Laboratory Animal
Co., Ltd. The C57BL/6 mice were housed under controlled conditions
at a temperature of 25±2˚C with a 12-h light-dark cycle and 55±2%
relative humidity, as previously described (32). Mice were and sacrificed by
intraperitoneal injection of overdoses of sodium pentobarbital (150
mg/kg). PDMs were cultured in an RPMI 1640 medium supplemented with
10% FBS, 100 U/ml penicillin and 100 µg/ml streptomycin in a
humidified atmosphere under 5% CO2 and 95% O2
at 37˚C. Cells were randomly separated into three groups: Normal
(N), LPS+ATP and AKFPD groups. PDMs in the LPS+ATP group were
stimulated with 500 ng/ml lipopolysaccharide (LPS; cat. no. L8880;
Beijing Solarbio Science & Technology Co., Ltd.) for 2.5 h,
followed by exposure to 5 mM ATP (cat. no. 10519979001; Roche
Diagnostics) for 0.5 h at 37˚C. PDMs in the AKFPD group were
pre-incubated with 400 µg/ml AKFPD for 24 h at 37˚C and
subsequently exposed to LPS plus ATP.
Immunofluorescence
The kidney tissue sections were fixed in 4%
paraformaldehyde for 24 h at 4˚C. The kidney tissue samples were
soaked in 70, 80 and 95% ethanol and anhydrous ethanol, followed by
embedding in paraffin and slicing into 4 µm-thick sections,
followed by permeabilization with 0.5% Triton X-100 in PBS for 20
min at 37˚C. After blocking with 5% bovine serum albumin (cat. no.
A8020; Solarbio) for 30 min at 37˚C, the sections were incubated
with primary antibodies against myeloperoxidase (MPO; 1:100
dilution; cat. no. 22225-1-AP; Proteintech Group, Inc.) overnight
at 4˚C. The slides were then incubated with goat anti-rabbit
secondary antibody (cat. no. AS007; ABclonal Biotech Co., Ltd.) at
37˚C for 45 min. The cell nuclei were counterstained with DAPI
(cat. no. KGA215-50; Nanjing KeyGen Biotech Co., Ltd.) for 3 min at
37˚C.
To detect colocalization using immunofluorescence,
kidney tissues or cells were separately incubated with anti-NLRP3
(1:100 dilution; cat. no. AG-20B-0006; AdipoGen Life Sciences),
anti-cathepsin B (1:150; cat. no. 12216-1-AP; Proteintech Group,
Inc.) and anti-ASC (1:200; cat. no. bs-6741R; Bioss Antibodies)
antibodies in a humidified chamber overnight at 4˚C, followed by
incubation with secondary antibodies Cy3 goat anti-rabbit IgG
(1:200; cat. no. As007; ABclonal) and Goat anti rabbit IgG/488
(1:100; cat. no. ZF-0511; ZSGB-BIO) for 45 min at 37˚C. The cell
nuclei were stained with DAPI. The immunostained samples were
visualized under a CKX53 confocal microscope (Olympus Corporation).
The quantitative changes were measured using Image-Pro Plus 6.0
(Media Cybernetics).
For lysosome detection, HK-2 cells and PDMs were
incubated with LysoTracker Red stain (cat. no. C1046; Beyotime
Institute of Biotechnology) for 1 h at 37˚C. After washing with PBS
three times, the cells were fixed with 4% paraformaldehyde for 10
min and stained with DAPI for 20 min at 37˚C. The cells were
examined using a confocal microscope (CKX53; Olympus Corp.) and
representative images were captured. The in vitro
experiments were performed in triplicate.
Cathepsin activity assay
After processing the cells and kidney tissues,
specific activity assay kits (cat. nos. ab65306, ab65307 and
ab65300; Abcam) were used to detect the activities of CTSL, CTSS
and CTSB, according to the manufacturer's instructions.
Fluorescence was quantified using an automatic microplate reader
(Beijing Liuyi Biotechnology Co. Ltd.) at excitation/emission
wavelengths of 400/505 nm.
Western blot analysis
The protein lysates from cultured cells and kidney
tissues were prepared as previously described (33), tissues and cells were lysed by RIPA
lysate buffer (cat. no. C1053; Applygen Technologies, Inc.) and
total protein was extracted. The protein content was determined
using a BCA protein assay kit (cat. no. E-BC-K318-M; Elabscience
Biotechnology, Inc.). Subsequently, 20-40 µg of total protein was
loaded per lane for SDS-PAGE. Proteins were separated by 8-15%
SDS-PAGE under reducing conditions and transferred onto PVDF
membranes (cat. no. IVPH00010; MilliporeSigma). The membranes were
blocked in 3% non-fat dry milk (cat. no. P1622; Beijing Pulilai
Gene Technology Co., Ltd.) in Tris-buffered saline with 0.1% Tween
20 detergent (TBS-T) at 37˚C for 1 h, followed by incubation with
primary antibodies overnight at 4˚C. The membranes were then
incubated with secondary antibodies at 37˚C for 2 h. ECL western
blotting detection reagent (Thermo Fisher Scientific, Inc.) was
used to visualize the protein bands, which were quantified using
the Tanon-5200 automatic chemiluminescence image analysis system
(Tanon Science and Technology Co., Ltd.). The intensity is
expressed as the relative protein expression, which was normalized
to the expression of β-actin. Image J software V1.8.0 (National
Institutes of Health) was adopted for the analysis of the grayscale
of the protein bands.
The following primary antibodies were used:
Anti-fibronectin (FN; 1:1,000 dilution; cat. no. ab268023; Abcam),
anti-α-smooth muscle actin (α-SMA; 1:1,000; cat. no. ab124964;
Abcam), anti-NLRP3 (1:1,000; cat. no. AG-20B-0006; AdipoGen Life
Sciences), anti-ASC (1:1,000; cat. no. bs-6741R; Bioss Antibodies),
anti-CTSB (1:500; cat. no. 12216-1-AP; Proteintech Group, Inc.),
anti-caspase-1 (1:1,000; cat. no. ab179515; Abcam), anti-IL-1β
(1:500; cat. no. sc-7884; Santa Cruz Biotechnology, Inc.) and
anti-β-actin (1:2,000; cat. no. TA-09; OriGene Technologies, Inc.).
The secondary horseradish peroxidase-conjugated antibodies used
were anti-rabbit IgG (1:2,000; cat. no. ZB-2301; ZSGB-BIO) and
anti-mouse IgG (1:2,000; cat. no. ZB-2305; ZSGB-BIO).
Statistical analysis
All experiments were performed at least three times,
and data are expressed as mean ± standard deviation for each group,
with the exception of the H&E scores, which were expressed
using median and interquartile ranges (IQR). Statistical analysis
was performed using IBM SPSS Statistics for Windows, version 19
(IBM Corp.). The Shapiro-Wilk normality test was used to test
normality. Comparisons between groups were conducted using a
one-way analysis of variance followed by Bonferroni analysis. For
analysis of H&E scores, the non-parametric Kruskal-Wallis
H-test was applied. P<0.05 was considered to indicate a
statistically significant difference.
Results
AKFPD ameliorates renal fibrosis in
UUO rats
As indicated in Fig.
1, H&E staining revealed that the kidney tissues of UUO
rats had the typical features of tubulointerstitial injury,
including tubular damage, inflammatory cell infiltration and
interstitial fibrosis (P<0.01; Fig.
1A and C). These changes were
attenuated by treatment with AKFPD on days 3 and 7 post-surgery
(P<0.01; Fig. 1A and C). Renal interstitial fibrosis evaluated
using Masson's trichrome staining indicated that the
tubulointerstitial fibrosis index of the UUO group was higher than
that of the sham group (P<0.01; Fig. 1B and D). However, the degree of renal
interstitial fibrosis was reduced by treatment with AKFPD on days 3
and 7 post-surgery (P<0.01; Fig.
1B and D).
 | Figure 1AKFPD ameliorates renal fibrosis in
the kidney tissue of UUO rats on days 3 and 7 after surgery. (A)
H&E staining of representative rat kidney tissue sections
(scale bar, 100 µm). (B) Masson's trichrome staining of
representative rat kidney tissue sections. (scale bar, 100 µm). (C)
H&E score for kidney damage. (D) The tubulointerstitial
fibrosis index is indicated. (E) Representative western blots of FN
and α-SMA in the rat kidney tissue. (F) Quantitative analysis of FN
and α-SMA protein expression in the rat kidney tissue. H&E
scores are expressed using median and interquartile range, with the
rest of the data are expressed as mean ± standard deviation (n=5
per group). *P<0.05 vs. sham group,
**P<0.01 vs. sham group, ##P<0.01 vs.
UUO group. AKFPD, fluorofenidone; UUO, unilateral ureteral
obstruction; H&E, hematoxylin-eosin; FN, fibronectin; α-SMA,
α-smooth muscle actin. |
Western blotting results indicated that the
expression of key fibrotic proteins (FN and α-SMA) was markedly
increased in the UUO group on days 3 and 7 post-surgery compared
with that in the sham group (P<0.01; Fig. 1E and F). Furthermore, the expression of
fibrotic proteins FN and α-SMA was decreased by treatment with
AKFPD (P<0.01; Fig. 1E and
F). The results showed that AKFPD
alleviated renal fibrosis in the UUO model.
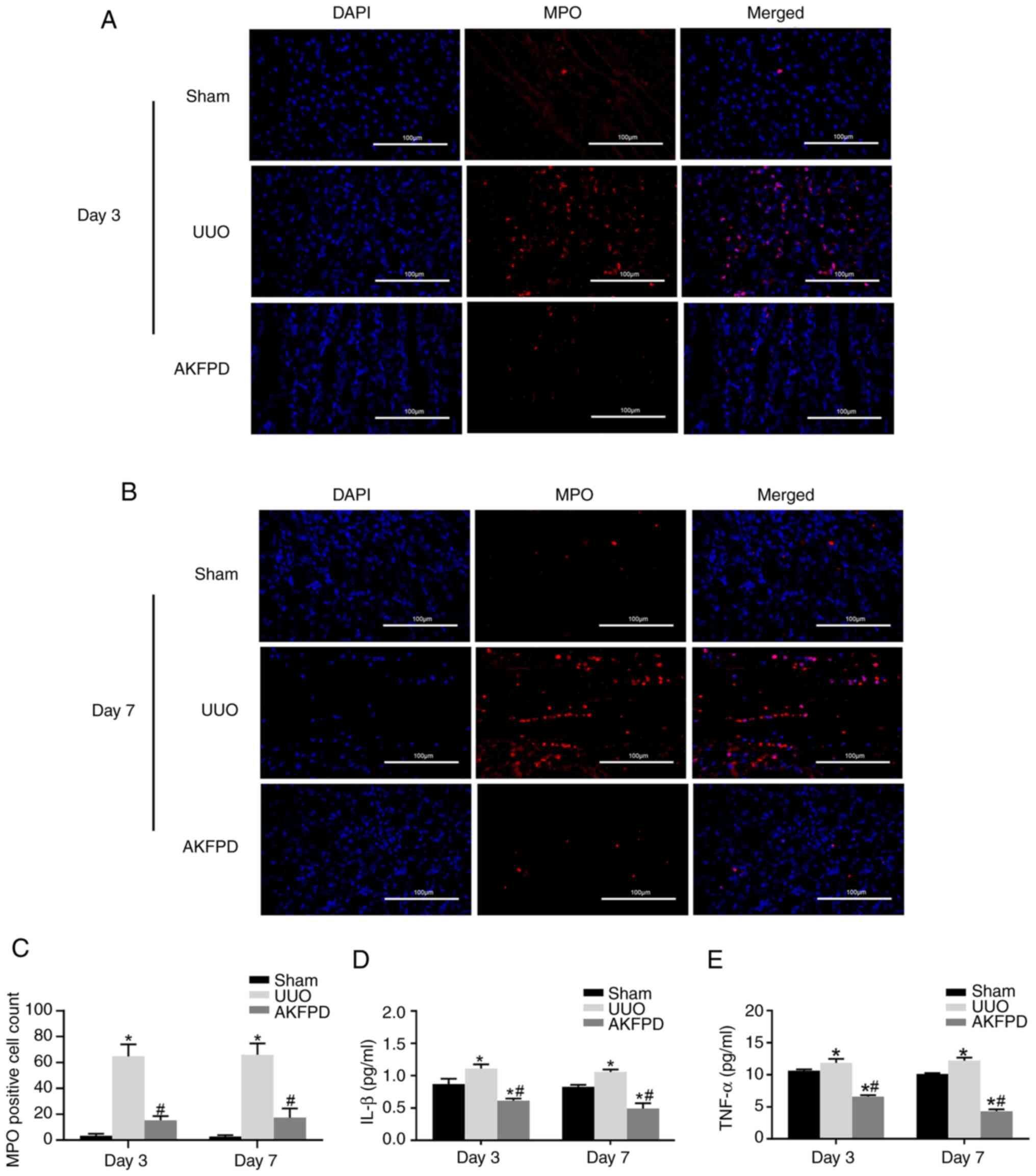
AKFPD suppresses renal inflammation in
UUO rats
Chronic renal inflammation promotes the occurrence
and progression of renal fibrosis. Renal inflammation is mainly
associated with inflammatory cytokines, such as IL-1β and TNF-α,
and infiltration of inflammatory cells, including neutrophils and
macrophages (34,35). To explore the effect of AKFPD on
renal inflammation, the abundance of MPO-positive cells in the
kidney tissue was determined using immunofluorescence (Fig. 2) because MPO is expressed in
activated macrophages, monocytes and neutrophils (36). MPO-positive cells were mainly
distributed around the renal tubules. As shown in Fig. 2A and C, the number of infiltrating MPO-positive
cells was significantly increased in the UUO group on days 3 and 7
post-surgery compared with that in the sham group. However, the
number of infiltrating MPO-positive cells decreased after treatment
with AKFPD (P<0.05).
Furthermore, the ELISA results revealed that the
expression of the inflammatory cytokines IL-1β and TNF-α was
significantly elevated in the UUO group on days 3 and 7
post-surgery compared with that in the sham group. However, these
elevated IL-1β and TNF-α levels decreased after treatment with
AKFPD treatment (P<0.05; Fig.
2D and E). In conclusion,
these results suggested that AKFPD reduces the expression of
inflammatory factors and the infiltration of inflammatory cells in
the UUO model.
AKFPD suppresses NLRP3 inflammasome
activation in UUO rats
NLRP3 inflammasome activation accelerates the
progression of renal fibrosis in UUO models (37,38).
In the present study, western blotting demonstrated that the
protein levels of NLRP3, ASC, pro-caspase-1, caspase-1, pro-IL-1β
and IL-1β were elevated in the UUO group on days 3 and 7
post-surgery compared with those in the sham group (Fig. 3). The protein levels of activated
caspase-1 and IL-1β were significantly reduced by treatment with
AKFPD (P<0.01; Fig. 3). There
was a slight increase in the NLRP3, ASC, pro-caspase-1 and
pro-IL-1β protein expression in the AKFPD group, but no
statistically significant difference with the UUO group
(P>0.05; Fig. 3). These
results suggested that AKFPD effectively downregulated activated
caspase-1 expression and IL-1β secretion by suppressing NLRP3
inflammasome activation.
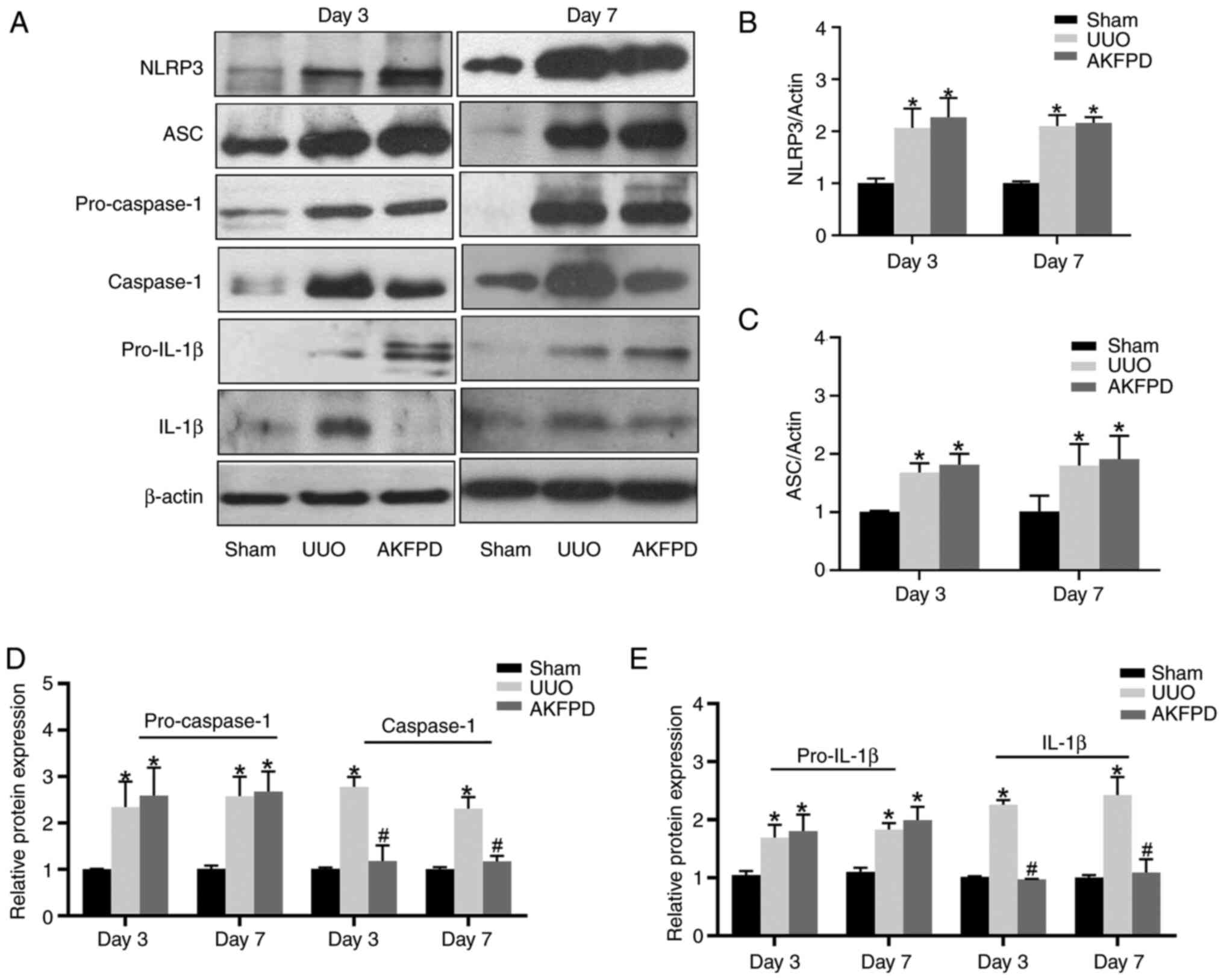 | Figure 3AKFPD inhibits NLRP3 inflammasome
activation in UUO rats. (A) Representative western blot of NLRP3,
ASC, pro-caspase-1, pro-IL-1β, caspase-1 and IL-1β expression in
the rat kidney tissues on days 3 and 7 post-surgery. (B-E)
Quantitative analysis of (B) NLRP3, pro-caspase-1, (C) ASC, (D)
pro-caspase-1 and caspase-1, and (E) pro-IL-1β and IL-1β protein
expression in rat kidney tissues. Data are presented as mean ±
standard deviation (n=5 per group). *P<0.05 vs. sham
group, #P<0.05 vs. UUO group. AKFPD, fluorofenidone;
NLRP3, NOD-like receptor thermal protein domain associated protein
3; UUO, unilateral ureteral obstruction; ASC, apoptosis-associated
speck-like protein containing a caspase recruitment domain. |
AKFPD inhibits NLRP3 inflammasome
activation in vitro
Macrophage-mediated inflammatory responses play an
integral role in the progression of kidney fibrosis (39). To investigate the mechanism by
which AKFPD suppresses NLRP3 inflammasome activation, LPS and ATP
were used to stimulate mouse PDMs to activate the NLRP3
inflammasome in vitro (Fig.
4). The western blotting results indicated that the protein
levels of NLRP3, ASC, pro-caspase-1, pro-IL-1β, caspase-1 and IL-1β
were substantially increased in stimulated PDMs compared with those
in normal cells; however, these elevated levels were significantly
decreased by treatment with AKFPD (P<0.05; Fig. 4A and B). Colocalization of NLRP3 with ASC in
the cytoplasm was increased in stimulated PDMs (reflected by
increased yellow immunofluorescent staining) compared with that in
normal cells (P<0.01; Fig. 4E
and G), whereas NLRP3-ASC binding
decreased by treatment with AKFPD (P<0.05; Fig. 4E and G). In addition to macrophages, NLRP3
inflammasome activation was investigated in HK-2 cells (40,41).
The protein levels of NLRP3, ASC, pro-caspase-1, caspase-1,
pro-IL-1β and IL-1β were increased in the H/R group compared with
those in the normal group (P<0.05; Fig. 4C and D) and these increases were reversed by
treatment with AKFPD (P<0.05; Fig.
4C and D). As shown in
Fig. 4F, confocal microscopy
demonstrated that the colocalization of NLRP3 with ASC was
remarkably increased in H/R-treated HK-2 cells (reflected by
increased yellow staining) compared with that in the normal group
(P<0.01; Fig. 4F and H) and this increase was rescued by
treatment with AKFPD (P<0.01; Fig.
4F and H). Overall, in
vitro results indicated that AKFPD reduced the production of
inflammatory factors by inhibiting NLRP3 inflammasome.
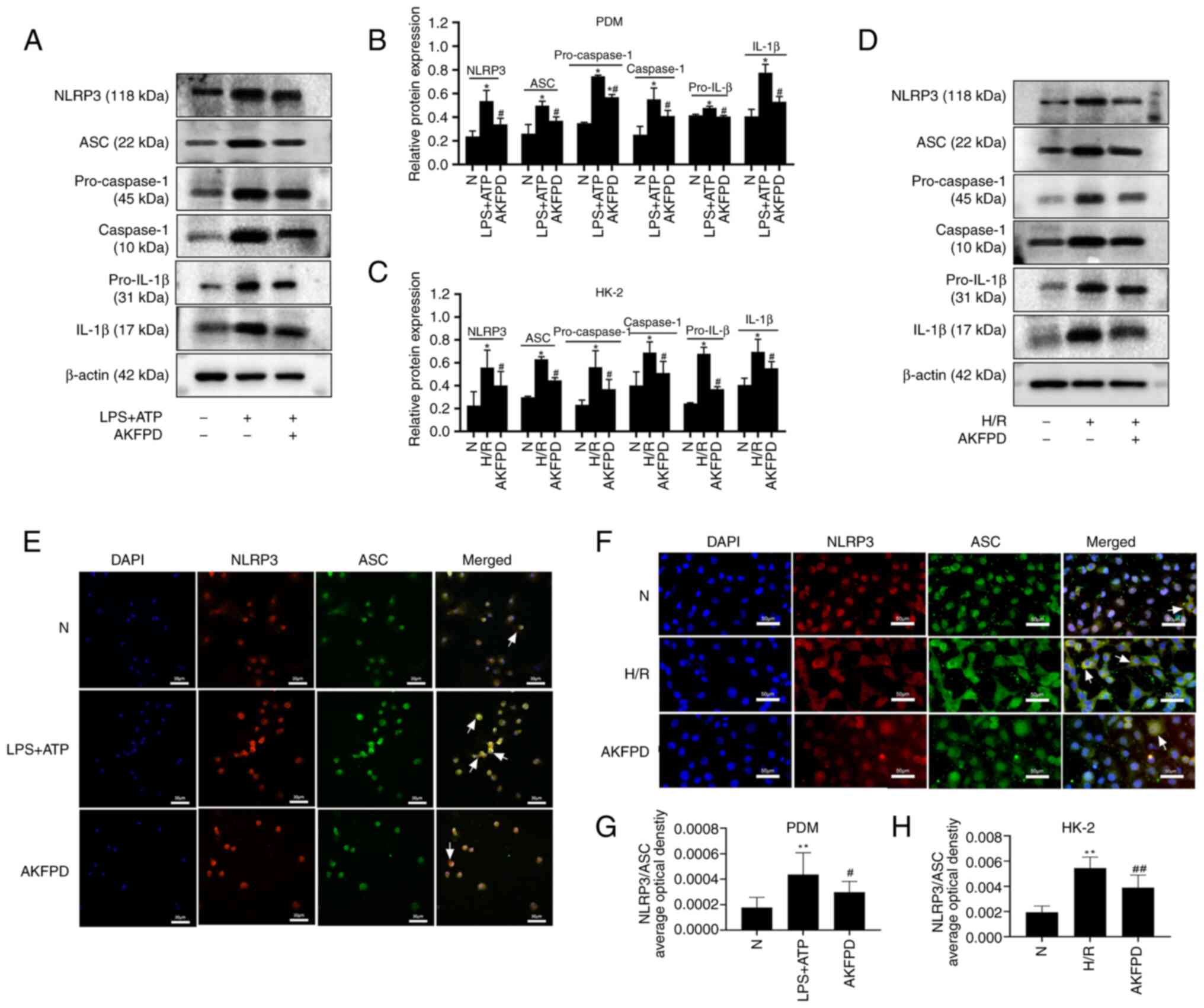 | Figure 4AKFPD inhibits NLRP3 inflammasome
activation in vitro. (A) Representative western blot and (B)
quantitative analysis of NLRP3, pro-IL-1β, caspase-1 and IL-1β
expression in LPS/ATP-stimulated PDMs. (C) Representative western
blot and (D) quantitative analysis of NLRP3, pro-IL-1β, caspase-1
and IL-1β expression in H/R-treated HK-2 cells. (E) Colocalization
of NLRP3 and ASC in PDMs visualized using immunofluorescent
staining. The white arrows indicate overlap (yellow).
(magnification, x400; scale bar, 30 µm). (F) Colocalization of
NLRP3 and ASC in HK-2 cells visualized using immunofluorescent
staining. The white arrows indicate overlap (yellow)
(magnification, x400; scale bar, 50 µm). (G) Quantification of the
immunofluorescent co-localization of NLRP3 and ASC in PDMs. The
quantitative changes were measured using Image-Pro Plus 6.0.
#P<0.05. (H) Quantification of the immunofluorescent
co-localization of NLRP3 and ASC in HK-2 cells. Data are presented
as the mean ± standard deviation (n=3 per group).
*P<0.05, **P<0.01 vs. N group;
#P<0.05 vs. H/R group or vs. LPS+ATP group,
##P<0.01 vs. H/R group. AKFPD, fluorofenidone; NLRP3,
NOD-like receptor thermal protein domain associated protein 3;
PDMs, peritoneal-derived macrophages; N, normal; H/R,
hypoxia/reoxygenation; ASC, apoptosis-associated speck-like protein
containing a caspase recruitment domain. |
AKFPD attenuates lysosomal cathepsin
activities in vitro
The NLRP3 inflammasome can be activated by danger
signals, including potassium efflux, lysosomal cathepsin leakage
and mtROS formation. Previous studies using cathepsin knockout or
knockdown models have revealed that multiple cathepsins are
involved in NLRP3 inflammasome activation (16,42).
Therefore, it was hypothesized that AKFPD suppressed NLRP3
inflammasome activation by downregulating lysosomal cathepsin
activity. As indicated in Fig. 5,
the cathepsin activity assay indicated that the activities of CTSB,
CTSS and CTSL were markedly elevated in LPS/ATP-stimulated PDMs and
H/R-treated HK-2 cells compared with those in normal cells, whereas
they were significantly decreased after treatment with AKFPD
(P<0.05; Fig. 5A and C). In addition, LysoTracker Red dye was
used to label acidic lysosomes, revealing that LPS/ATP-stimulated
PDMs and H/R-treated HK-2 cells exhibited significantly increased
numbers of lysosomes or induced lysosome aggregation compared with
those in normal cells (P<0.01; Fig.
5B and D-F), and this effect
was inhibited after treatment with AKFPD (P<0.01; Fig. 5B and D-F). The results showed that AKFPD
protected lysosomal damage and downregulated lysosomal cathepsins
activity in vitro.
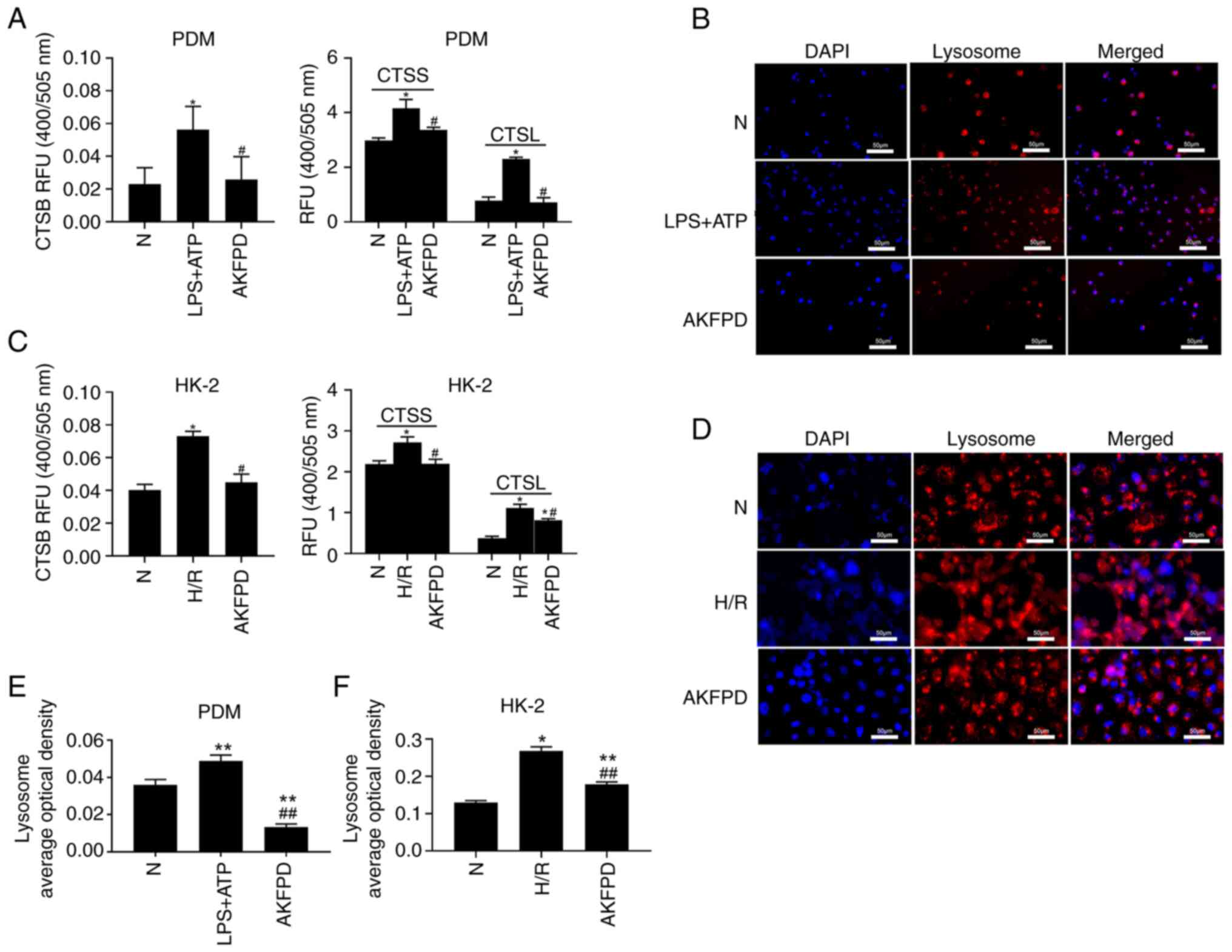 | Figure 5AKFPD downregulates cathepsin
activity in vitro. (A) Effects of AKFPD on cathepsin (CTSB,
CTSS and CTSL) activities in PDMs. (B) Effects of AKFPD on the
accumulation of lysosomes visualized using LysoTracker Red dye in
PDMs (magnification, x400; scale bar, 50 µm). (C) Effects of AKFPD
on cathepsin (CTSB, CTSS and CTSL) activities in HK-2 cells. (D)
Effects of AKFPD on the accumulation of lysosomes visualized using
LysoTracker Red dye in HK-2 cells (magnification, x400; scale bar,
50 µm). (E) Quantitative analysis of the effect of AKFPD on
lysosome accumulation visualized in PDMs using LysoTracker red dye.
(F) Quantitative analysis of the effect of AKFPD on lysosome
accumulation visualized in HK-2 cells using LysoTracker red dye.
Data are presented as mean ± standard deviation (n=3 per group).
*P<0.05, **P<0.01 vs. N group;
#P<0.05 ##P<0.01 vs. H/R group or vs.
LPS + ATP group. AKFPD, fluorofenidone; CTSB, cathepsin B; CTSS,
cathepsin S; CTSL cathepsin L; N, normal; PDMs, peritoneal-derived
macrophages; H/R, hypoxia/reoxygenation. |
AKFPD suppresses NLRP3 inflammasome
activation by decreasing cathepsin B expression in vitro
CTSB is required for IL-1β production and is induced
by a diverse set of NLRP3 inflammasome activators (43). Lysosomal cathepsins, especially
CTSB, activate the NLRP3 inflammasome via direct interactions
(44). As presented in Fig. 6, in the present study, western
blotting indicated that the CTSB protein level was significantly
increased in LPS/ATP-stimulated PDMs compared with that in normal
cells (P<0.05; Fig. 6A), and
the same results were observed in H/R-treated HK-2 cells
(P<0.05; Fig. 6D). However,
treatment with AKFPD decreased the elevated CTSB protein level
in vitro (P<0.05; Fig.
6A and D). The expression and
colocalization of CTSB with NLRP3 were evaluated in vitro
using immunofluorescence for further validation. The immunostaining
results indicated a low association between CTSB and NLRP3 in
normal cells. By contrast, LPS/ATP-stimulated PDMs and H/R-treated
HK-2 cells exhibited increased colocalization of CTSB with NLRP3
compared with that in normal cells (P<0.01; Fig. 6B, C, E and
F). These results suggested that
CTSB was released from lysosomes and colocalized with NLRP3 in the
cytoplasm under inflammatory conditions induced by LPS/ATP
stimulation or H/R treatment. As predicted, these changes were
abrogated by treatment with AKFPD (P<0.05; Fig. 6B, C, E and
F). These findings support the
hypothesis that AKFPD inhibited NLRP3 inflammasome activation by
reducing the expression of CTSB protein in vitro.
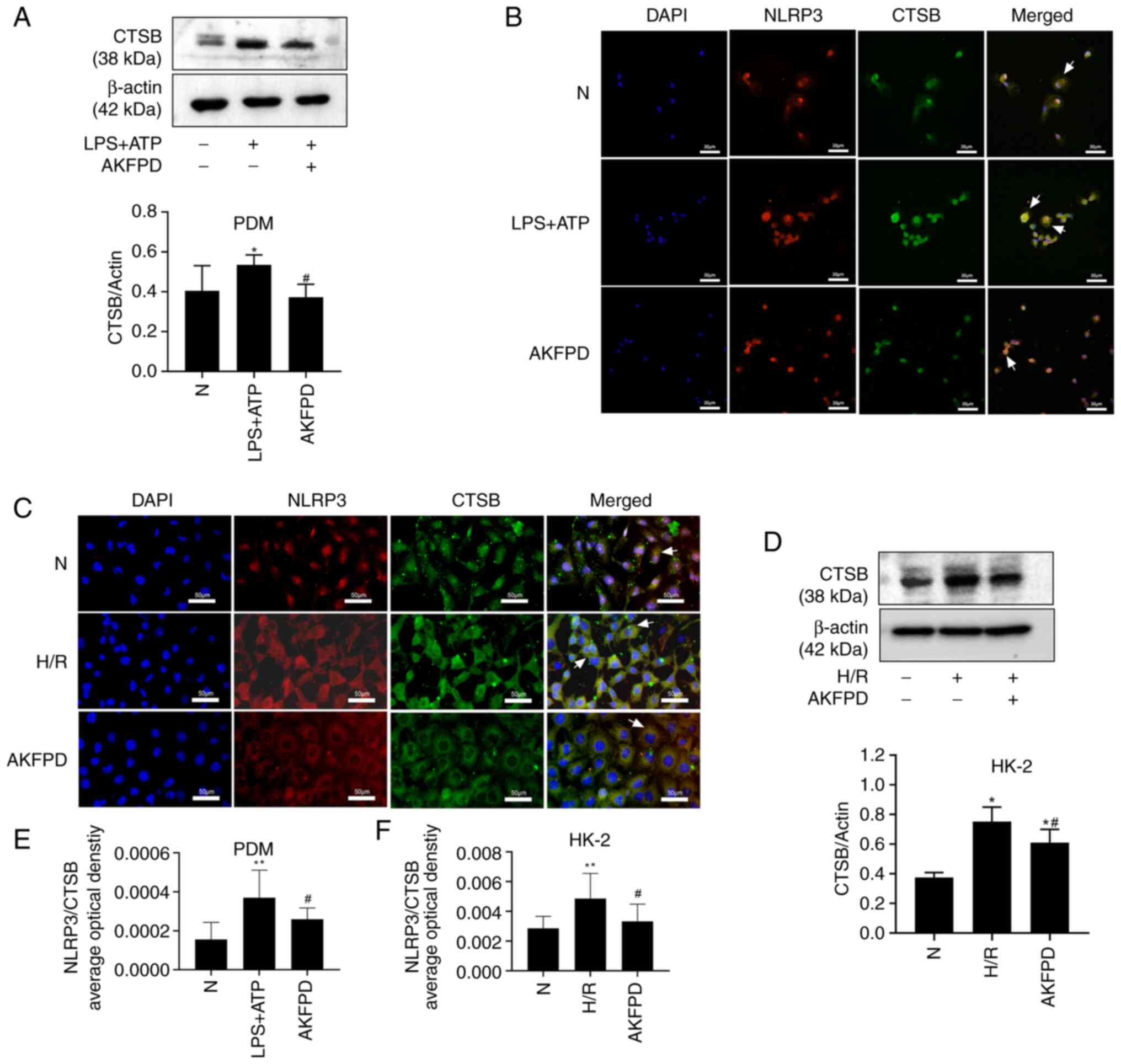 | Figure 6AKFPD inhibits NLRP3 inflammasome
activation by CTSB expression in vitro. (A) Representative
western blot and quantitative data of CTSB in LPS + ATP-stimulated
PDMs. (B) Effects of AKFPD on the colocalization of NLRP3 and CTSB
in activated PDMs visualized using immunofluorescent staining.
White arrows indicate overlap (yellow) (magnification, x400; scale
bar, 30 µm). (C) Effects of AKFPD on the colocalization of CTSB and
NLRP3 in H/R-treated HK-2 cells visualized using immunofluorescent
staining. White arrows represent overlap (yellow) (magnification,
x400; scale bar, 50 µm). (D) Representative western blot and
quantitative data of CTSB in H/R-treated HK-2 cells. (E)
Quantitative analysis of the effect of AKFPD on NLRP3 and CTSB
co-localization in activated PDM visualized using immunofluorescent
staining. The quantitative changes were measured using Image-Pro
Plus 6.0. (F) Quantitative analysis of the effect of AKFPD on the
co-localization of CTSB and NLRP3 in H/R-treated HK-2 cells
observed using immunofluorescent staining. The quantitative changes
were measured using Image-Pro Plus 6.0. Data are presented as mean
± standard deviation (n=3 per group). *P<0.05,
**P<0.01 vs. N group; #P<0.05 vs. H/R
group or vs. LPS + ATP group. AKFPD, fluorofenidone; NLRP3,
NOD-like receptor thermal protein domain associated protein 3;
CTSB, cathepsin B; PDMs, peritoneal-derived macrophages; N, normal;
H/R, hypoxia/reoxygenation. |
AKFPD inhibits NLRP3 inflammasome
activation via the lysosome pathway in UUO rats
To further confirm the mechanism underlying the
aforementioned results, AKFPD suppression of cathepsin activity was
investigated in UUO rats (Fig. 7).
The activities of CTSB, CTSS and CTSL were significantly increased
in the UUO group compared with those in the sham group on days 3
and 7 post-surgery (P<0.05; Fig.
7A) and were significantly decreased after treatment with AKFPD
(P<0.05; Fig. 7A). Western
blotting indicated that the CTSB protein expression level was
elevated on days 3 and 7 post-surgery in the UUO group compared
with that in the sham group. However, treatment with AKFPD reduced
this increased CTSB expression level (P<0.05; Fig. 7C and E). Subsequently, the immunofluorescence
results indicated that the colocalization of CTSB with NLRP3
sharply increased in the UUO group on days 3 and 7 post-surgery
compared with that in the sham group. However, it was significantly
decreased after treatment with AKFPD (Fig. 7B and D). Taken together, the results suggest
that AKFPD suppressed NLRP3 inflammasome activation via the
lysosome pathway in vivo.
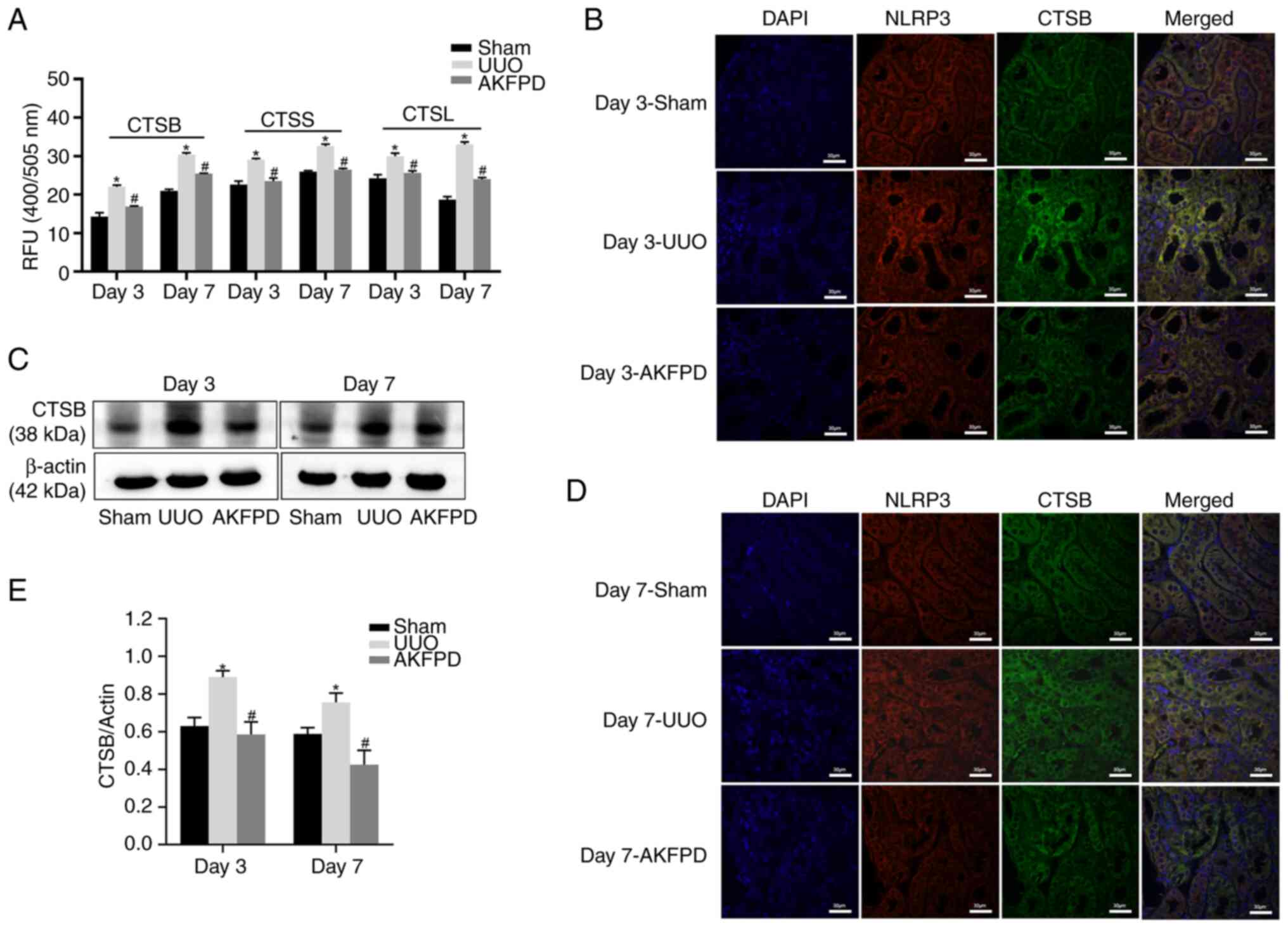 | Figure 7AKFPD inhibits CTSB-mediated NLRP3
inflammasome activation in UUO rats. (A) Effects of AKFPD on
cathepsin (CTSB, CTSS and CTSL) activities in the rat kidney
tissues. (B) Effects of AKFPD on the colocalization of NLRP3 and
CTSB in the rat kidney tissues visualized using immunostaining in
UUO rats on day 3 (magnification, x400; scale bar, 30 µm). (C)
Representative western blot of CTSB in the rat kidney tissues. (D)
Effects of AKFPD on the colocalization of NLRP3 and CTSB in the rat
kidney tissues visualized using immunostaining in UUO rats on day 7
(magnification, x400; scale bar, 30 µm). (E) Quantitative data of
CTSB in the rat kidney tissues. Data are presented as mean ±
standard deviation (n=5 per group). *P<0.05 vs. sham
group, #P<0.05 vs. UUO group. AKFPD, fluorofenidone;
CTSB, cathepsin B; NLRP3, NOD-like receptor thermal protein domain
associated protein 3; UUO, unilateral ureteral obstruction; CTSS,
cathepsin S; CTSL cathepsin L. |
Discussion
The final characteristic feature of end-stage kidney
disease is renal fibrosis (45),
for which there is no effective clinical treatment. Chronic renal
inflammation is an important contributor to chronic kidney disease.
Renal inflammation is caused by macrophage accumulation and
inflammatory cell infiltration in kidney tissues (35). Consistent with the findings of
previous studies (46,47), the results of the present study
indicated that AKFPD suppressed extracellular matrix deposition,
decreased interstitial infiltration of inflammatory cells and
downregulated inflammatory cytokine production in the kidney
tissues of UUO rats. Furthermore, the results suggested that AKFPD
attenuated renal fibrosis by suppressing leached lysosomal
cathepsins, thereby further inhibiting the activation of the
downstream cathepsin-NLRP3 inflammasome pathway in UUO rats. These
findings provided new insights into a previously unknown molecular
mechanism underlying the protective effects of AKFPD against renal
fibrosis.
Emerging evidence indicates that the NLRP3
inflammasome, which comprises NLRP3, ASC and pro-caspase-1,
promotes the development of renal fibrosis (48-50).
The NLRP3 inflammasome acts as an important danger-recognition
platform and can be activated by pathogen-associated molecular
patterns and damage-associated molecular patterns (51). These two steps of initiation and
activation are necessary for NLRP3 inflammasome activation. The
initiation step (signal 1) is related to the NF-κB pathway and
pro-IL-1β production whereas the activation step (signal 2)
contributes to the oligomerization of the NLRP3 inflammasome and
caspase-1-mediated secretion of IL-1β (52). The results of the present study
demonstrated that the protein expression of NLRP3 inflammasome
components was elevated and activated under renal fibrosis
conditions both in vivo and in vitro. Furthermore,
treatment with AKFPD significantly reduced the expression of
activated caspase-1 and secretion of IL-1β in vivo,
indicating that NLRP3 inflammasome activation was significantly
inhibited. However, AKFPD did not affect the protein levels of the
NLRP3 inflammasome components NLRP3, ASC and pro-caspase-1. These
results suggested that the inhibition of NLRP3 inflammasome
activation underlies the anti-inflammatory effects of AKFPD in UUO
rats. Notably, treatment with AKFPD significantly reduced the
expression of NLRP3, ASC and pro-caspase-1 in vitro,
suggesting it may affect the NF-κB pathway of the initiation signal
(signal 1) in vitro. Additionally, AKFPD treatment also
suppressed NLRP3 inflammasome activation in activated PDMs and
H/R-treated HK-2 cells. Overall, the results suggest a common
mechanism in vivo and in vitro by which AKFPD
specifically suppresses NLRP3 inflammasome activation via the
activation step (signal 2).
Lysosomal cysteine cathepsins are a family of
enzymes that require an acidic, reducing environment for their
activation. Previous studies have reported that multiple cathepsins
exert a positive effect on NLRP3 inflammasome activation (17,53).
siRNA knockdown of CTSB, CTSL and CTSS
decreases the levels of caspase-1 and IL-1β in active PDMs and
H/R-treated HK-2 cells, suggesting that NLRP3 inflammasome
activation was inhibited (42).
Another study demonstrated that lysosomal cathepsin expression is
significantly elevated in UUO mice, suggesting that pharmacological
blocking of cathepsin activity may help treat kidney fibrosis
(14). In the present study, the
activities of lysosomal CTSB, CTSS and CTSL were increased in
stimulated cells in which the NLRP3 inflammasome was activated but
were notably decreased by treatment with AKFPD. The effect of AKFPD
on the cathepsin-NLRP3 activation pathway was further confirmed in
UUO rats, in which lysosomal disruption resulted in the release of
cathepsins into the cytoplasm of kidney tissues; the cathepsins
were then involved in NLRP3 inflammasome activation. Consistent
with the in vitro experiment results, treatment with AKFPD
downregulated cathepsin activity in vivo. A number of
studies have shown that lysosomal damage causes lysosomal membrane
destabilization and releases large amounts of CTSB into the
cytoplasm, which plays a critical role in NLRP3 inflammasome
activation and is required for caspase-1 activation and IL-1β
production (54,55). As CTSB plays a critical role in
NLRP3 inflammasome activation, the present study focused on the
effect of AKFPD on the activation of the NLRP3 inflammasome by
CTSB. The findings indicated that AKFPD plays a role in suppressing
the expression of CTSB, thereby inhibiting its involvement in NLRP3
inflammasome activation. Therefore, the present study provided
novel insights into the mechanism by which AKFPD inhibits NLRP3
inflammasome activation.
In the present study, the abundance of lysosomes
increased upon stimulation in vitro and was significantly
decreased by treatment with AKFPD. As previously reported, the
formation of autolysosomes contributes to the elimination of
damaged lysosomes (5). Therefore,
these results suggested that AKFPD may also enhance the
autophagic-lysosomal degradation pathway to eliminate damaged
lysosomes. Furthermore, increasing evidence suggests that autophagy
reduces the expression of NLRP3 inflammasome-associated cytokines,
thereby suppressing inflammation (56). These findings indicate a new
mechanism by which AKFPD inhibits inflammation via the suppression
of NLRP3 inflammasome activation.
The present study had some limitations, including
the lack of a comprehensive investigation of the underlying
mechanisms using knockout or overexpression experiments.
The present study confirmed that AKFPD alleviates
renal fibrosis by suppressing lysosomal cathepsin-mediated
activation of the NLRP3 inflammasome. However, further studies are
required to confirm whether AKFPD affects the autolysosome
pathway.
Acknowledgements
The authors would like to thank Professor Lijian Tao
(Department of Nephrology, Xiangya Hospital of Central South
University, Changsha, China) for providing AKFPD.
Funding
Funding: The present study was supported by the National Natural
Science Foundation of China (grant no. 81960678) and the Natural
Science Foundation of Jiangxi Province (grant no.
20181BAB215010).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
LZhe, YY and XW designed the present study. LZhe,
WM, ZH, XL and WL performed the experiments. LZha, JZ, QW and JL
analyzed the data. LZhe and YY wrote the manuscript. LZhe and YY
confirm the authenticity of all the raw data. All authors have read
and approved the final version of the manuscript.
Ethics approval and consent to
participate
The present study protocol was approved by the
Medical Research Ethics Committee of the First Affiliated Hospital
of Nanchang University (approval no. 2020-1-59).
Patient consent for publication
Not applicable.
Competing interests
The authors declare they have no competing
interests.
References
|
1
|
Hackl MJ, Burford JL, Villanueva K, Lam L,
Suszták K, Schermer B, Benzing T and Peti-Peterdi J: Tracking the
fate of glomerular epithelial cells in vivo using serial
multiphoton imaging in new mouse models with fluorescent lineage
tags. Nat Med. 19:1661–1666. 2013.PubMed/NCBI View
Article : Google Scholar
|
|
2
|
Lovisa S, LeBleu VS, Tampe B, Sugimoto H,
Vadnagara K, Carstens JL, Wu CC, Hagos Y, Burckhardt BC,
Pentcheva-Hoang T, et al: Epithelial-to-mesenchymal transition
induces cell cycle arrest and parenchymal damage in renal fibrosis.
Nat Med. 21:998–1009. 2015.PubMed/NCBI View
Article : Google Scholar
|
|
3
|
Li X, Pan J, Li H, Li G, Liu X, Liu B, He
Z, Peng Z, Zhang H, Li Y, et al: DsbA-L mediated renal
tubulointerstitial fibrosis in UUO mice. Nat Commun.
11(4467)2020.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Meng XM, Nikolic-Paterson DJ and Lan HY:
Inflammatory processes in renal fibrosis. Nat Rev Nephrol.
10:493–503. 2014.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Kimura T, Isaka Y and Yoshimori T:
Autophagy and kidney inflammation. Autophagy. 13:997–1003.
2017.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Jo EK, Kim JK, Shin DM and Sasakawa C:
Molecular mechanisms regulating NLRP3 inflammasome activation. Cell
Mol Immunol. 13:148–159. 2016.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Alyaseer AAA, de Lima MHS and Braga TT:
The role of NLRP3 inflammasome activation in the epithelial to
mesenchymal transition process during the fibrosis. Front Immunol.
11(883)2020.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Song H, Zhao C, Yu Z, Li Q, Yan R, Qin Y,
Jia M and Zhao W: UAF1 deubiquitinase complexes facilitate NLRP3
inflammasome activation by promoting NLRP3 expression. Nat Commun.
11(6042)2020.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Bakker PJ, Butter LM, Claessen N, Teske
GJ, Sutterwala FS, Florquin S and Leemans JC: A tissue-specific
role for Nlrp3 in tubular epithelial repair after renal
ischemia/reperfusion. Am J Pathol. 184:2013–2022. 2014.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Vilaysane A, Chun J, Seamone ME, Wang W,
Chin R, Hirota S, Li Y, Clark SA, Tschopp J, Trpkov K, et al: The
NLRP3 inflammasome promotes renal inflammation and contributes to
CKD. J Am Soc Nephrol. 21:1732–1744. 2010.PubMed/NCBI View Article : Google Scholar
|
|
11
|
He H, Jiang H, Chen Y, Ye J, Wang A, Wang
C, Liu Q, Liang G, Deng X, Jiang W and Zhou R: Oridonin is a
covalent NLRP3 inhibitor with strong anti-inflammasome activity.
Nat Commun. 9(2550)2018.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Kim SM, Kim YG, Kim DJ, Park SH, Jeong KH,
Lee YH, Lim SJ, Lee SH and Moon JY: Inflammasome-independent role
of NLRP3 mediates mitochondrial regulation in renal injury. Front
Immunol. 9(2563)2018.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Cocchiaro P, De Pasquale V, Della Morte R,
Tafuri S, Avallone L, Pizard A, Moles A and Pavone LM: The
multifaceted role of the lysosomal protease cathepsins in kidney
disease. Front Cell Dev Biol. 5(114)2017.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Fox C, Cocchiaro P, Oakley F, Howarth R,
Callaghan K, Leslie J, Luli S, Wood KM, Genovese F, Sheerin NS and
Moles A: Inhibition of lysosomal protease cathepsin D reduces renal
fibrosis in murine chronic kidney disease. Sci Rep.
6(20101)2016.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Musante L, Tataruch D, Gu D, Liu X,
Forsblom C, Groop PH and Holthofer H: Proteases and protease
inhibitors of urinary extracellular vesicles in diabetic
nephropathy. J Diabetes Res. 2015(289734)2015.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Tang TT, Lv LL, Pan MM, Wen Y, Wang B, Li
ZL, Wu M, Wang FM, Crowley SD and Liu BC: Hydroxychloroquine
attenuates renal ischemia/reperfusion injury by inhibiting
cathepsin mediated NLRP3 inflammasome activation. Cell Death Dis.
9(351)2018.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Allan ERO, Campden RI, Ewanchuk BW, Tailor
P, Balce DR, McKenna NT, Greene CJ, Warren AL, Reinheckel T and
Yates RM: A role for cathepsin Z in neuroinflammation provides
mechanistic support for an epigenetic risk factor in multiple
sclerosis. J Neuroinflammation. 14(103)2017.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Meng J, Zou Y, Hu C, Zhu Y, Peng Z, Hu G,
Wang Z and Tao L: Fluorofenidone attenuates bleomycin-induced
pulmonary inflammation and fibrosis in mice via restoring caveolin
1 expression and inhibiting mitogen-activated protein kinase
signaling pathway. Shock. 38:567–573. 2012.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Peng ZZ, Hu GY, Shen H, Wang L, Ning WB,
Xie YY, Wang NS, Li BX, Tang YT and Tao LJ: Fluorofenidone
attenuates collagen I and transforming growth factor-beta1
expression through a nicotinamide adenine dinucleotide phosphate
oxidase-dependent way in NRK-52E cells. Nephrology (Carlton).
14:565–572. 2009.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Peng Y, Yang H, Zhu T, Zhao M, Deng Y, Liu
B, Shen H, Hu G, Wang Z and Tao L: The antihepatic fibrotic effects
of fluorofenidone via MAPK signalling pathways. Eur J Clin Invest.
43:358–368. 2013.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Ning WB, Hu GY, Peng ZZ, Wang L, Wang W,
Chen JY, Zheng X, Li J and Tao LJ: Fluorofenidone inhibits Ang
II-induced apoptosis of renal tubular cells through blockage of the
Fas/FasL pathway. Int Immunopharmacol. 11:1327–1332.
2011.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Tu S, Jiang Y, Cheng H, Yuan X, He Y, Peng
Y, Peng X, Peng Z, Tao L and Yang H: Fluorofenidone protects liver
against inflammation and fibrosis by blocking the activation of
NF-κB pathway. FASEB J. 35(e21497)2021.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Tang Y, Zhang F, Huang L, Yuan Q, Qin J,
Li B, Wang N, Xie Y, Wang L, Wang W, et al: The protective
mechanism of fluorofenidone in renal interstitial inflammation and
fibrosis. Am J Med Sci. 350:195–203. 2015.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Kocak MZ, Aktas G, Atak BM, Duman TT, Yis
OM, Erkus E and Savli H: Is Neuregulin-4 a predictive marker of
microvascular complications in type 2 diabetes mellitus? Eur J Clin
Invest. 50(e13206)2020.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Bilgin S, Kurtkulagi O, Atak BM, Duman TT,
Kahveci G, Khalid A and Aktas G: Does C-reactive protein to serum
albumin ratio correlate with diabetic nephropathy in patients with
type 2 diabetes mellitus? The care time study. Prim Care Diabetes.
15:1071–1074. 2021.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Aktas G, Yilmaz S, Kantarci DB, Duman TT,
Bilgin S, Balci SB and Atak BM: Is serum uric acid-to-HDL
cholesterol ratio elevation associated with diabetic kidney injury?
Postgrad Med. 135:519–523. 2023.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Taslamacioglu Duman T, Ozkul FN and Balci
B: Could systemic inflammatory index predict diabetic kidney injury
in type 2 diabetes mellitus? Diagnostics (Basel).
13(2063)2023.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Zheng L, Zhang J, Yuan X, Tang J, Qiu S,
Peng Z, Yuan Q, Xie Y, Mei W, Tang Y, et al: Fluorofenidone
attenuates interleukin-1β production by interacting with NLRP3
inflammasome in unilateral ureteral obstruction. Nephrology
(Carlton). 23:573–584. 2018.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Liao X, Jiang Y, Dai Q, Yu Y, Zhang Y, Hu
G, Meng J, Xie Y, Peng Z and Tao L: Fluorofenidone attenuates renal
fibrosis by inhibiting the mtROS-NLRP3 pathway in a murine model of
folic acid nephropathy. Biochem Biophys Res Commun. 534:694–701.
2021.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Chevalier RL, Forbes MS and Thornhill BA:
Ureteral obstruction as a model of renal interstitial fibrosis and
obstructive nephropathy. Kidney Int. 75:1145–1152. 2009.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Lu M, Li H, Liu W, Zhang X, Li L and Zhou
H: Curcumin attenuates renal interstitial fibrosis by regulating
autophagy and retaining mitochondrial function in unilateral
ureteral obstruction rats. Basic Clin Pharmacol Toxicol.
128:594–604. 2021.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Lu M, Yang W, Peng Z, Zhang J, Mei W, Liu
C, Tang J, Ma H, Yuan X, Meng J, et al: Fluorofenidone inhibits
macrophage IL-1β production by suppressing inflammasome activity.
Int Immunopharmacol. 27:148–153. 2015.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Zhang J, Zheng L, Yuan X, Liu C, Yuan Q,
Xie F, Qiu S, Peng Z, Tang Y, Meng J, et al: Mefunidone ameliorates
renal inflammation and tubulointerstitial fibrosis via suppression
of IKKβ phosphorylation. Int J Biochem Cell Biol. 80:109–118.
2016.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Xiang H, Zhu F, Xu Z and Xiong J: Role of
inflammasomes in kidney diseases via both canonical and
non-canonical pathways. Front Cell Dev Biol. 8(106)2020.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Imig JD and Ryan MJ: Immune and
inflammatory role in renal disease. Compr Physiol. 3:957–976.
2013.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Jiang Y, Quan J, Chen Y, Liao X, Dai Q, Lu
R, Yu Y, Hu G, Li Q, Meng J, et al: Fluorofenidone protects against
acute kidney injury. FASEB J. 33:14325–14336. 2019.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Seo JB, Choi YK, Woo HI, Jung YA, Lee S,
Lee S, Park M, Lee IK, Jung GS and Park KG: Gemigliptin attenuates
renal fibrosis through down-regulation of the NLRP3 inflammasome.
Diabetes Metab J. 43:830–839. 2019.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Zheng Z, Xu K, Li C, Qi C, Fang Y, Zhu N,
Bao J, Zhao Z, Yu Q, Wu H and Liu J: NLRP3 associated with chronic
kidney disease progression after ischemia/reperfusion-induced acute
kidney injury. Cell Death Discov. 7(324)2021.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Bhatia D, Chung KP, Nakahira K, Patino E,
Rice MC, Torres LK, Muthukumar T, Choi AM, Akchurin OM and Choi ME:
Mitophagy-dependent macrophage reprogramming protects against
kidney fibrosis. JCI Insight. 4(e132826)2019.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Lorenz G, Darisipudi MN and Anders HJ:
Canonical and non-canonical effects of the NLRP3 inflammasome in
kidney inflammation and fibrosis. Nephrol Dial Transplant.
29:41–48. 2014.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Conley SM, Abais JM, Boini KM and Li PL:
Inflammasome activation in chronic glomerular diseases. Curr Drug
Targets. 18:1019–1029. 2017.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Orlowski GM, Colbert JD, Sharma S, Bogyo
M, Robertson SA and Rock KL: Multiple cathepsins promote Pro-IL-1β
synthesis and NLRP3-Mediated IL-1β activation. J Immunol.
195:1685–1697. 2015.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Kelley N, Jeltema D, Duan Y and He Y: The
NLRP3 inflammasome: An overview of mechanisms of activation and
regulation. Int J Mol Sci. 20(3328)2019.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Martine P and Rébé C: Heat shock proteins
and inflammasomes. Int J Mol Sci. 20(4508)2019.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Bozic M, Caus M, Rodrigues-Diez RR,
Pedraza N, Ruiz-Ortega M, Garí E, Gallel P, Panadés MJ, Martinez A,
Fernández E and Valdivielso JM: Protective role of renal proximal
tubular alpha-synuclein in the pathogenesis of kidney fibrosis. Nat
Commun. 11(1943)2020.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Dai Q, Zhang Y, Liao X, Jiang Y, Lv X,
Yuan X, Meng J, Xie Y, Peng Z, Yuan Q, et al: Fluorofenidone
alleviates renal fibrosis by inhibiting necroptosis through
RIPK3/MLKL pathway. Front Pharmacol. 11(534775)2020.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Yang H, Zhang W, Xie T, Wang X and Ning W:
Fluorofenidone inhibits apoptosis of renal tubular epithelial cells
in rats with renal interstitial fibrosis. Braz J Med Biol Res.
52(e8772)2019.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Wu M, Han W, Song S, Du Y, Liu C, Chen N,
Wu H, Shi Y and Duan H: NLRP3 deficiency ameliorates renal
inflammation and fibrosis in diabetic mice. Mol Cell Endocrinol.
478:115–125. 2018.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Mulay SR: Multifactorial functions of the
inflammasome component NLRP3 in pathogenesis of chronic kidney
diseases. Kidney Int. 96:58–66. 2019.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Li L, Tang W and Yi F: Role of
inflammasome in chronic kidney disease. Adv Exp Med Biol.
1165:407–421. 2019.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Lamkanfi M: Emerging inflammasome effector
mechanisms. Nat Rev Immunol. 11:213–220. 2011.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Man SM and Kanneganti TD: Regulation of
inflammasome activation. Immunol Rev. 265:6–21. 2015.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Terada K, Yamada J, Hayashi Y, Wu Z,
Uchiyama Y, Peters C and Nakanishi H: Involvement of cathepsin B in
the processing and secretion of interleukin-1beta in chromogranin
A-stimulated microglia. Glia. 58:114–124. 2010.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Stancu IC, Cremers N, Vanrusselt H,
Couturier J, Vanoosthuyse A, Kessels S, Lodder C, Brône B, Huaux F,
Octave JN, et al: Aggregated Tau activates NLRP3-ASC inflammasome
exacerbating exogenously seeded and non-exogenously seeded Tau
pathology in vivo. Acta Neuropathol. 137:599–617. 2019.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Chevriaux A, Pilot T, Derangere V, Simonin
H, Martine P, Chalmin F, Ghiringhelli F and Rébé C: Cathepsin B is
required for NLRP3 inflammasome activation in macrophages, through
NLRP3 interaction. Front Cell Dev Biol. 8(167)2020.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Nakahira K, Haspel JA, Rathinam VA, Lee
SJ, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim
HP, et al: Autophagy proteins regulate innate immune responses by
inhibiting the release of mitochondrial DNA mediated by the NALP3
inflammasome. Nat Immunol. 12:222–230. 2011.PubMed/NCBI View Article : Google Scholar
|