Introduction
Hepatocellular carcinoma (HCC) is the sixth most
common malignant tumor and the third leading cause of
cancer-related mortality worldwide (1,2). Due
to lack of clear symptoms in the early stages of HCC, the majority
of patients with HCC have advanced stage cancer upon diagnosis,
with a 5-year survival rate of <20% (3-5).
Even after curative resection, the prognosis of patients with HCC
remains unsatisfactory owing to the biological characteristics of
HCC cells, including metastasis and recurrence (6). Therefore, identifying the molecular
basis underlying HCC and exploring potential molecular targets for
HCC therapy are of great urgency.
Growth differentiation factor 11 (GDF11), also known
as bone morphogenetic protein (BMP)11, is a member of the
transforming growth factor-β superfamily and BMP subfamily
(7). Evidence has indicated that
GDF11 serves as a critical modulator in tumor progression. GDF11
acts as a tumor suppressor in triple-negative breast cancer by
preserving epithelial cell-cell adhesion and inhibiting cell
invasion (8). Furthermore,
colorectal cancer-associated human intestinal lymphatic endothelial
cells have been reported to promote tumor cell proliferation via
the soluble matrisome component GDF11(9). Exosome-transmitted microRNA-3124-5p
promotes cholangiocarcinoma development by targeting GDF11(10). GDF11 overexpression in pancreatic
cancer cells suppresses the proliferation, migration and invasion
abilities in vitro (11).
In addition, it has been reported that GDF11 suppresses
adipogenesis and improves the metabolic functioning of mature
adipocytes via the WNT/β-catenin and ALK5/SMAD2/3 pathways
(12). GDF11 is also involved in
metabolic reprogramming and lipid metabolism dysregulation in HCC
cells through ALK5-dependent signaling (13). Notably, GDF11 has been verified to
be lowly expressed in liver cancer tissues and cell lines compared
with that in normal liver tissues and cells (14), and GDF11 exerts tumor-suppressive
properties in HCC cells by restricting proliferation,
clonogenicity, spheroid formation and cellular function
(15). However, the intrinsic mechanisms underlying the
antitumor effects of GDF11 in HCC have not yet been fully
elucidated.
GDF11 has been suggested to serve as a mammalian
target of rapamycin (mTOR) inhibitory factor in HCC cells (16). mTOR, a serine/threonine protein
kinase, exists as two structurally and functionally different
complexes, known as mTOR complex (mTORC)1 and mTORC2 (17,18).
mTORC1 can modulate cell proliferation and metabolism and can
suppress autophagy (19).
Autophagy is critical for maintaining cellular homeostasis and
survival. By contrast, inhibition of mTORC1 activates autophagy,
which is required for the clearance of dysfunctional cellular
components (20).
In the present study, the biological role of GDF11
in the malignant behavior of HCC cells was assessed. Moreover,
whether GDF11 could exert antitumor effects against HCC via
mediating the mTORC1-autophagy axis was discussed.
Materials and methods
Cell culture
The human normal hepatocyte cell line HHL-5 and the
human hepatoma cell line Huh-7 were purchased from Shanghai
Enzyme-linked Biotechnology Co., Ltd. The human hepatoma cell lines
SNU-449 and Hep3B, and the immortalized hybrid human umbilical vein
endothelial cell (HUVEC)/EAhy926 cell line were purchased from the
American Type Culture Collection. All cells were cultured in DMEM
supplemented with 10% FBS and 1% penicillin-streptomycin (all from
Gibco; Thermo Fisher Scientific, Inc.) at 37˚C in a 5%
CO2 incubator.
Cell transfection
The GDF11 overexpression plasmid (Oe-GDF11), which
was established by inserting the GDF11 gene into the pcDNA3.1
vector, and the empty vector [Oe-negative control (NC)] were
designed by Shanghai GenePharma Co., Ltd. Cell transfection with
either Oe-GDF11 or Oe-NC was performed using
Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.). Briefly, Oe-GDF11 or Oe-NC (4 µg) and
Lipofectamine 2000 (10 µl) were added to Opti-MEM (250 µl; Gibco;
Thermo Fisher Scientific, Inc.) and incubated for 10 min at room
temperature. Subsequently, diluted vectors were mixed with diluted
Lipofectamine 2000 and then incubated for 15 min at room
temperature. Huh-7 cells were re-plated in serum-free DMEM, and the
transfection mixtures were separately added to the cells when the
cell confluence reached 85% for 4 h of incubation at 37˚C. After
another 48 h of incubation in complete DMEM at 37˚C, the cells were
collected for subsequent experiments.
Cell treatment
Huh-7 hepatoma cells were divided into the control
group (cells cultured under normal conditions), the Oe-NC group
(cells transfected with Oe-NC), the Oe-GDF11 group (cells
transfected with Oe-GDF11) and the Oe-GDF11 + MHY1485 group [cells
transfected with Oe-GDF11 and treated with 10 µM mTOR activator
MHY1485 (MedChemExpress) for 4 h at 37˚C].
Western blotting
Total protein from HHL-5, SNU-449, Hep3B and Huh-7
cells was extracted using RIPA lysis buffer (Beyotime Institute of
Biotechnology) containing protease inhibitors, and the protein
concentration was quantified using the BCA method. Protein samples
(30 µg/lane) were separated by 5-10% SDS-PAGE and transferred to
PVDF membranes. After blocking in 5% BSA (Thermo Fisher Scientific,
Inc.) for 2 h at 37˚C, the membranes were incubated on a shaker
with primary antibodies against GDF11 (1:5,000; cat. no. ab234647),
phosphorylated (p)-mTOR (1:1,000; cat. no. ab109268), mTOR
(1:10,000; cat. no. ab134903), p-p70 S6K T389 (1:1,000; cat. no.
ab2571), p70 S6K (1:1,000; cat. no. ab308113), p-S6 (1:5,000; cat.
no. ab215214), S6 (1:1,000; cat. no. ab127980), LC3-II/I (1:2,000;
cat. no. ab192890), Beclin-1 (1:2,000; cat. no. ab207612), p62
(1:10,000; cat. no. ab109012), Bcl-2 (1:2,000; cat. no. ab182858),
Bax (1:10,000; cat. no. ab32503), cleaved caspase-3 (1:500; cat.
no. ab32042), caspase-3 (1:5,000; cat. no. ab32351), CDK4
(1:10,000; cat. no. ab108357), CDK6 (1:50,000; cat. no. ab124821),
cyclin D1 (1:200 dilution; cat. no. ab16663), E-cadherin (1:25;
cat. no. ab227639), N-cadherin (1:5,000; cat. no. ab76011), Snail
(1:1,000; cat. no. ab216347), Vimentin (1:5,000; cat. no. ab92547),
GAPDH (1:2,500; cat. no. ab9485) and β-actin (1:5,000; cat. no.
ab8227) (all from Abcam) overnight at 4˚C. Subsequently, the
membranes were incubated with HRP-conjugated secondary antibodies
(1:20,000; cat. no. ab6721; Abcam) for 1 h at room temperature.
GAPDH served as the internal controls. Protein bands were
visualized using an ECL detection kit (Beyotime Institute of
Biotechnology) and were semi-quantified using ImageJ (version 1.42;
National Institutes of Health).
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA from HHL-5, SNU-449, Hep3B and Huh-7 cells
was extracted using TRIzol® (Invitrogen; Thermo Fisher
Scientific, Inc.) and was then reverse-transcribed into cDNA using
the Prime Script RT reagent kit (Takara Bio, Inc.) according to the
manufacturer's protocol. qPCR was carried out using the SYBR Green
PCR Kit (Takara Bio, Inc.) on an ABI Detection System (PerkinElmer,
Inc.). The qPCR thermocycling conditions were as follows: 95˚C for
10 min, followed by 40 cycles of 95˚C for 15 sec and 64˚C for 30
sec. GAPDH served as the internal control. The following primers
were employed: GDF11, 5'-CCACCACCGAGACCGTCATT-3' (forward) and
5'-GAGGGCTGCCATCTGTCTGT-3' (reverse); GAPDH,
5'-GCACCGTCAAGGCTGAGAAC-3' (forward) and 5'-ATGGTGGTGAAGACGCCAGT-3'
(reverse). The mRNA expression levels of GDF11 were calculated
using the 2-ΔΔCq method (21).
Immunofluorescence (IF) staining
After fixation in 4% paraformaldehyde for 30 min at
room temperature, Huh-7 cells were permeabilized with 0.1% Triton
X-100 for 15 min and then blocked with 5% BSA (Beyotime Institute
of Biotechnology) for 2 h at room temperature. Subsequently, the
cells were incubated with an anti-LC3 primary antibody (1:250; cat.
no. ab225382, Abcam) overnight at 4˚C and were then incubated with
a FITC-conjugated secondary antibody (1:1,000; cat. no. ab150077,
Abcam) for 1 h at room temperature. DAPI was used to counterstain
the nuclei for 5 min in the dark at room temperature. Fluorescence
images were captured under a fluorescence microscope
(magnification, x200).
Cell viability assay
Cell viability was investigated using a Cell
Counting Kit-8 (CCK-8) assay (Beyotime Institute of Biotechnology).
Huh-7 cells (5,000 cells/well) grown in a 96-well plate were
cultured for 24, 48 and 72 h. Subsequently, 10 µl CCK-8 reagent was
added to each well for an additional 2 h of incubation. The optical
density value was measured at 450 nm using a microplate reader
(Bio-Rad Laboratories, Inc.).
EdU proliferation assay
Cell proliferative ability was investigated using a
commercial EdU cell proliferation kit (Beyotime Institute of
Biotechnology). Huh-7 cells were incubated with EdU reaction
cocktail for 2 h at 37˚C to complete EdU labeling. The cells were
then fixed in 4% paraformaldehyde for 30 min and permeabilized with
0.5% Triton X-100 for 20 min at room temperature. DAPI was applied
to counterstain the nuclei for 5 min in the dark at room
temperature. Fluorescence images were captured under a fluorescence
microscope (magnification, x100).
Colony formation assay
Cell colony-forming ability was investigated using a
colony formation assay. Huh-7 cells (600 cells/well) grown in a
6-well plate were continuously cultured in 5% CO2 at
37˚C for 14 days. Cell colonies were fixed in 4% paraformaldehyde
for 30 min and stained with 0.1% crystal violet for 15 min at room
temperature. Images of visible colonies (≥50 cells) were captured
under a light microscope and colonies were counted using ImageJ
(version 1.42).
Flow cytometry
For cell apoptosis analysis, Huh-7 cells were
trypsinized, collected by centrifugation at 1,000 x g for 5 min at
room temperature, resuspended in 300 µl binding buffer at a
concentration of 1x106 cells/ml, and doubly stained with
5 µl Annexin V-FITC and PI (Beyotime Institute of Biotechnology) in
the dark for 15 min at room temperature. The cells were then
subjected to BD FACSCanto™ flow cytometry (FACSCalibur;
BD Biosciences) and analyzed using FlowJo™ software
(version 10.8.1; FlowJo LLC) to assess cell apoptosis. For cell
cycle analysis, Huh-7 cells (5x105) were trypsinized,
collected by centrifugation at 1,000 x g for 5 min at room
temperature, resuspended in PBS and then fixed in 70% ice-cold
ethanol at 4˚C for 4 h. Thereafter, the cells were stained with 1
ml PI/RNase dye (50 µg/ml) for 30 min at room temperature in the
dark and were subjected to BD FACSCanto™ flow cytometry
(FACSCalibur; BD Biosciences) and analyzed using FlowJo™
software (version 10.8.1; FlowJo LLC) to assess cell cycle
distribution.
Wound healing assay
Cell migration was investigated using a wound
healing assay. Huh-7 cells were grown in a 6-well plate until 95%
confluence. The cell monolayer was scraped with a 200-µl sterile
pipette tip to create a wound, followed by a 24-h incubation in
serum-free DMEM at 37˚C. Images of the wounds were captured at 0
and 24 h under a light microscope (magnification, x100).
Transwell assay
Cell invasion ability was investigated using a
Transwell invasion assay. Huh-7 cells suspended in fresh serum-free
DMEM at a density of 2x104 cells were seeded into the
upper chamber of Transwell plates (8-µm pore size; Costar; Corning,
Inc.) precoated with Matrigel at 37˚C for 30 min. DMEM supplemented
with 10% FBS was added to the lower chamber to serve as a
chemoattractant. After a 24-h incubation at 37˚C, non-migrated
cells in the upper chamber were removed with cotton swabs and cells
that had invaded into the lower chamber were fixed with 4%
paraformaldehyde for 30 min at room temperature, stained with 0.5%
crystal violet for 10 min at room temperature and captured under a
light microscope (magnification, x200).
Tube formation
The conditioned media (CM) of normal Huh-7 cells,
Huh-7 cells transfected with Oe-NC, Huh-7 cells transfected with
Oe-GDF11, and Huh-7 cells transfected with Oe-GDF11 and treated
with MHY1485 were collected after ~24 h incubation at 37˚C. HUVECs
(2x104 cells/well) seeded in 96-well plates precoated
with Matrigel were cultured in the collected CM at 37˚C for 24 h.
Images of tube formation were captured under a light microscope
(magnification, x100).
Statistical analysis
Data analysis was performed using GraphPad Prism
(version 9.1; GraphPad Software; Dotmatics). Experimental data from
three independent repeats are presented as the mean ± standard
deviation. Comparisons among multiple groups were performed using
one-way analysis of variance followed by Tukey's post hoc test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
GDF11 strengthens autophagy in HCC
cells by suppressing the mTORC1 signaling pathway
Differences in the expression levels of GDF11 in
HHL-5 human normal hepatocytes and the human hepatoma cell lines
SNU-449, Hep3B and Huh-7 were investigated by RT-qPCR and western
blotting. In comparison with HHL-5 cells, GDF11 mRNA and protein
expression levels were markedly downregulated in HCC cells,
particularly in Huh-7 cells (Fig.
1A). Thus, Huh-7 cells were selected for subsequent research.
Huh-7 cells were transfected with Oe-GDF11 or Oe-NC for functional
experiments and transfection with Oe-GDF11 significantly
upregulated GDF11 expression compared with the Oe-NC group
(Fig. 1B). In addition, IF
staining revealed that GDF11 overexpression increased LC3
accumulation compared with that in the Oe-NC group (Fig. 1C). Furthermore, the expression
levels of genes in the mTORC1-autophagy axis were analyzed. GDF11
overexpression decreased the expression levels of p-mTOR, p-p70 S6K
T389, p-S6 and p62, and increased the expression levels of
LC3-II/LC3-I and Beclin-1 compared with those in the Oe-NC group
(Fig. 1D). These findings
indicated that GDF11 overexpression suppressed the mTORC1 signaling
pathway and enhanced autophagy in Huh-7 cells. Moreover, the
enhancement of LC3 accumulation caused by GDF11 overexpression was
partially reversed by treatment with mTOR activator MHY1485
(Fig. 2A). Decreased p-mTOR, p-p70
S6K T389, p-S6 and p62 protein expression levels, as well as
increased LC3-II/LC3-I and Beclin-1 protein expression levels
induced by GDF11 overexpression were partially reversed by MHY1485
treatment (Fig. 2B). These results
indicated that GDF11 could strengthen autophagy in HCC cells by
suppressing the mTORC1 signaling pathway.
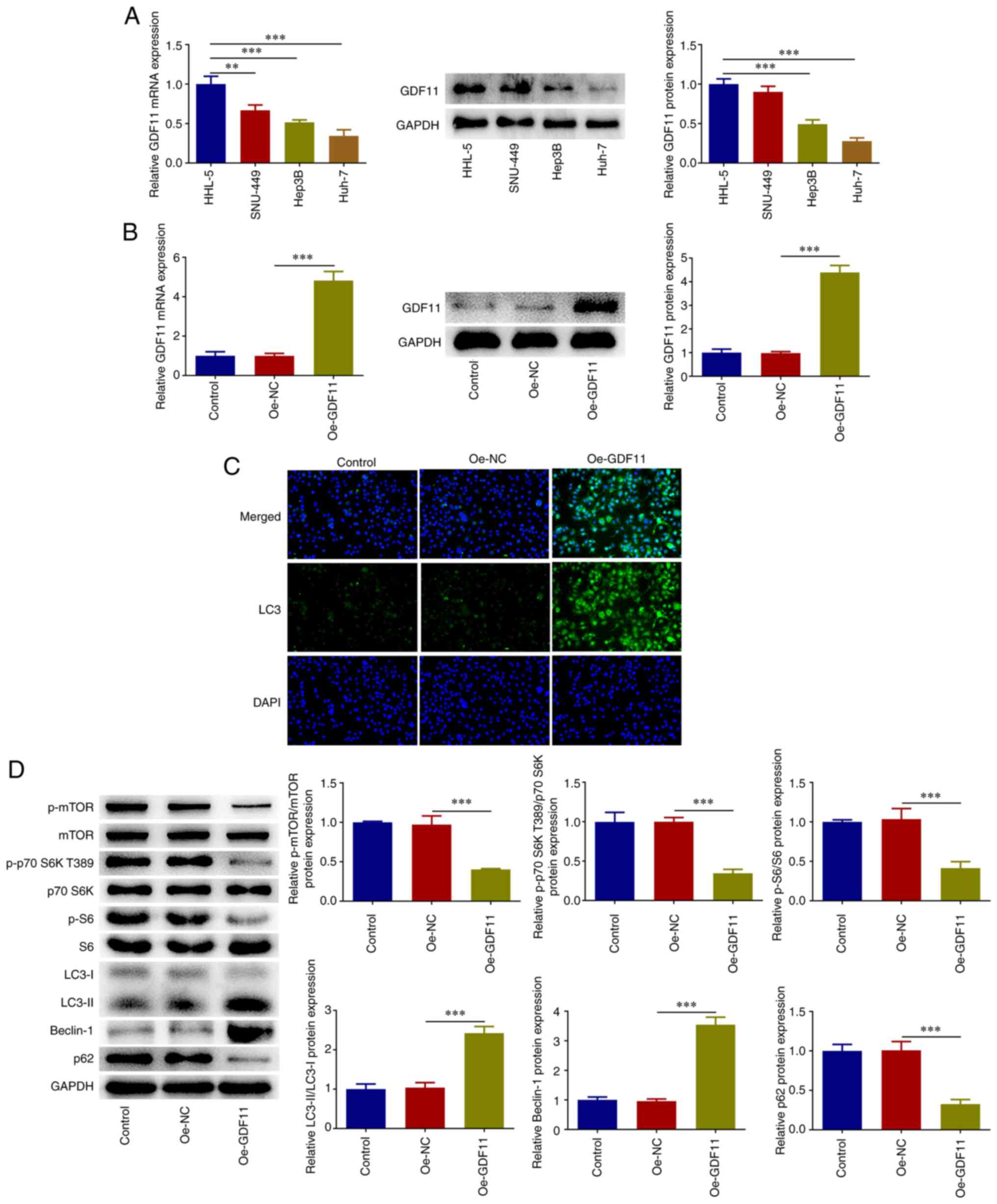 | Figure 1GDF11 suppresses the mTORC1 signaling
pathway and strengthens autophagy in hepatocellular carcinoma
cells. (A) Differences in GDF11 expression in HHL-5 human normal
hepatocytes and the human hepatoma cell lines SNU-449, Hep3B and
Huh-7 were detected by RT-qPCR and western blotting. (B) Huh-7
cells were transfected with either Oe-GDF11 or Oe-NC, and
transfection efficiency was validated by RT-qPCR and western
blotting. (C) Huh-7 cells were transfected with either Oe-GDF11 or
Oe-NC. LC3 expression was determined by immunofluorescence
staining. (D) Huh-7 cells were transfected with either Oe-GDF11 or
Oe-NC. p-mTOR, mTOR, p-p70 S6K T389, p70 S6K, p-S6, S6, LC3-I,
LC3-II, Beclin-1 and p62 protein expression levels were detected by
western blotting. **P<0.01, ***P<0.001.
GDF11, growth differentiation factor 11; mTORC1, mammalian target
of rapamycin complex 1; RT-qPCR, reverse transcription-quantitative
PCR; Oe, overexpression plasmid; NC, negative control; p,
phosphorylated. |
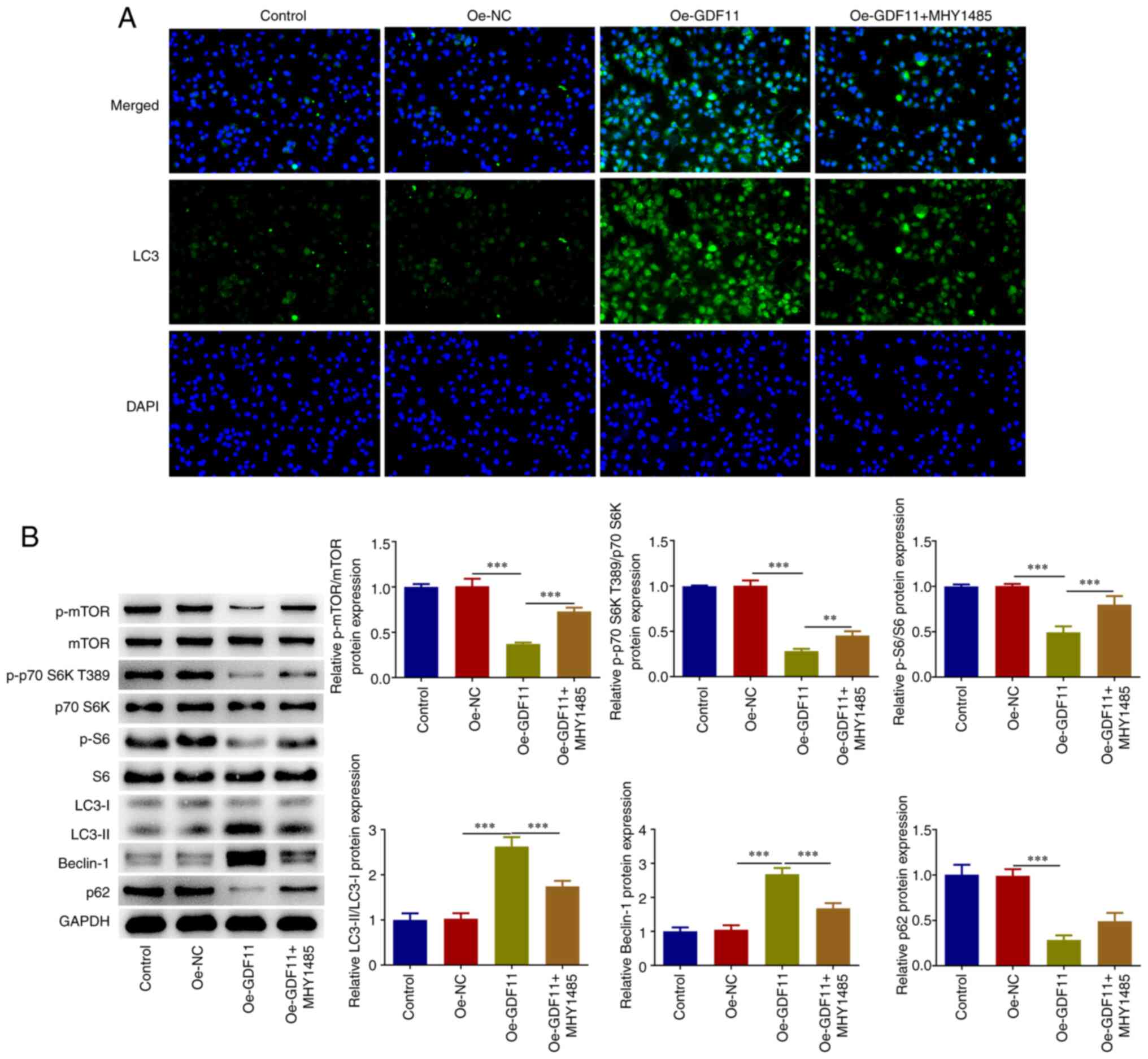 | Figure 2GDF11 strengthens autophagy in
hepatocellular carcinoma cells by suppressing the mTORC1 signaling
pathway. Huh-7 cells were transfected with either Oe-GDF11 or
Oe-NC. Oe-GDF11-transfected Huh-7 cells were treated with the mTOR
activator MHY1485. (A) LC3 expression was determined by
immunofluorescence staining. (B) p-mTOR, mTOR, p-p70 S6K T389, p70
S6K, p-S6, S6, LC3-I, LC3-II, Beclin-1 and p62 protein expression
levels were detected by western blotting. **P<0.01,
***P<0.001. GDF11, growth differentiation factor 11;
mTORC1, mammalian target of rapamycin complex 1; Oe, overexpression
plasmid; NC, negative control; p, phosphorylated. |
GDF11 inhibits the proliferation and
colony-forming ability of HCC cells by suppressing the mTORC1
signaling pathway
The results of the CCK-8 assay indicated that GDF11
overexpression suppressed proliferation of Huh-7 cells compared
with that in the Oe-NC group, whereas MHY1485 treatment partially
restored the impaired cell proliferative capacity (Fig. 3A). As determined by EdU staining,
GDF11 overexpression suppressed cell proliferation compared with in
the Oe-NC group, as indicated by the reduced number of
EdU+ Huh-7 cells, and this was partially reversed by
MHY1485 treatment (Fig. 3B).
Furthermore, the colony formation assay revealed that GDF11
overexpression suppressed the colony formation of Huh-7 cells
compared with that in the Oe-NC group, which was partially reversed
by MHY1485 treatment (Fig. 3C).
These findings suggested that GDF11 could inhibit the proliferation
and colony-forming ability of HCC cells by suppressing the mTORC1
signaling pathway.
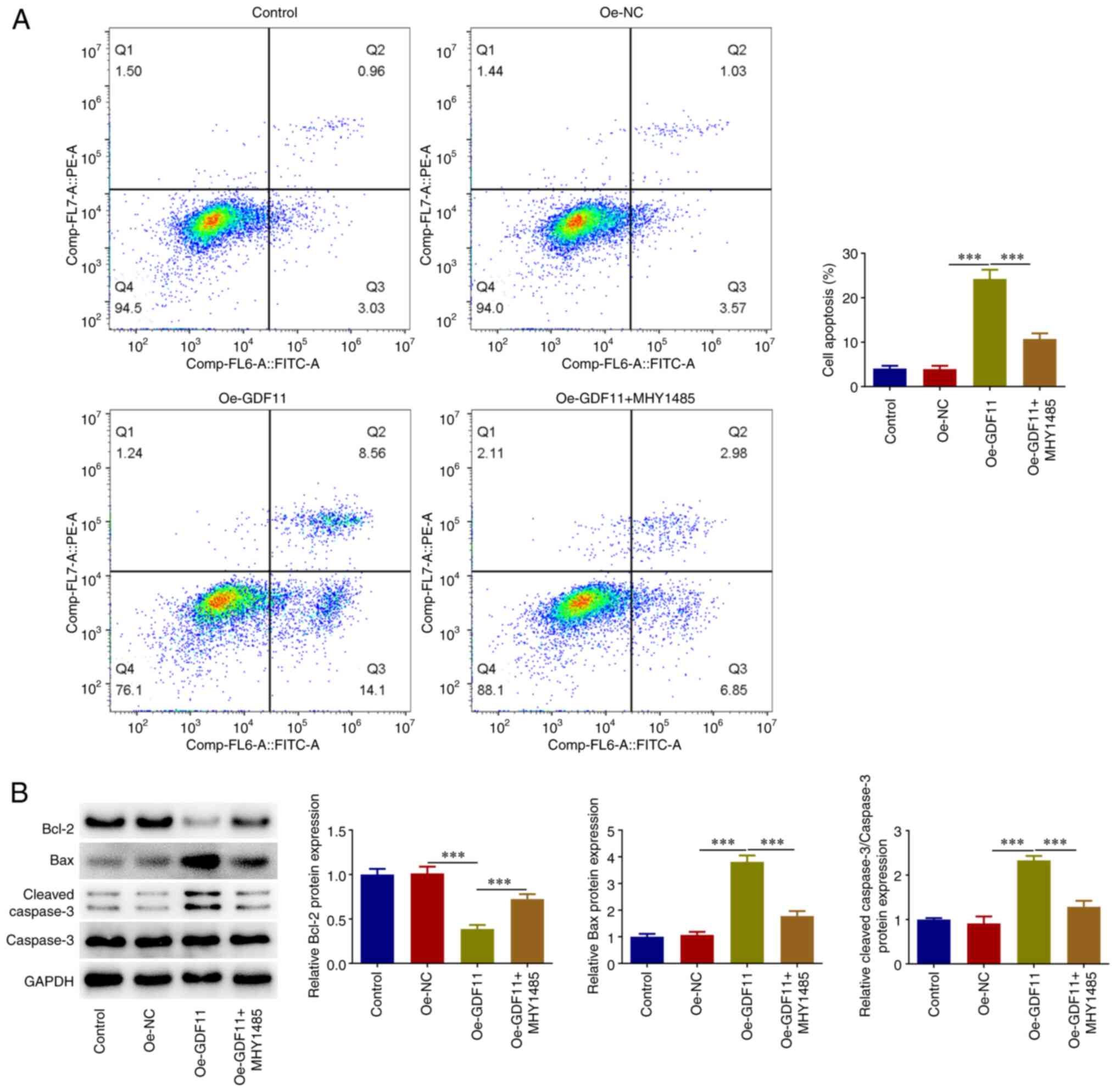
GDF11 facilitates HCC cell apoptosis
by suppressing the mTORC1 signaling pathway
Flow cytometry was used for the cell apoptosis
analysis. Apoptotic rate was calculated as the sum of the early
apoptosis rate and the late apoptosis rate. It was revealed that
GDF11 overexpression elevated the apoptotic rate of Huh-7 cells
compared with that in the Oe-NC group, which was partially reversed
by MHY1485 treatment (Fig. 4A).
Additionally, GDF11 overexpression reduced Bcl-2 protein expression
levels, and elevated Bax and cleaved caspase-3 protein expression
levels compared with the Oe-NC group. By contrast, MHY1485
treatment partially reversed the regulatory effects of GDF11
overexpression on apoptosis-associated proteins (Fig. 4B). These results suggested that
GDF11 may facilitate HCC cell apoptosis by suppressing the mTORC1
signaling pathway.
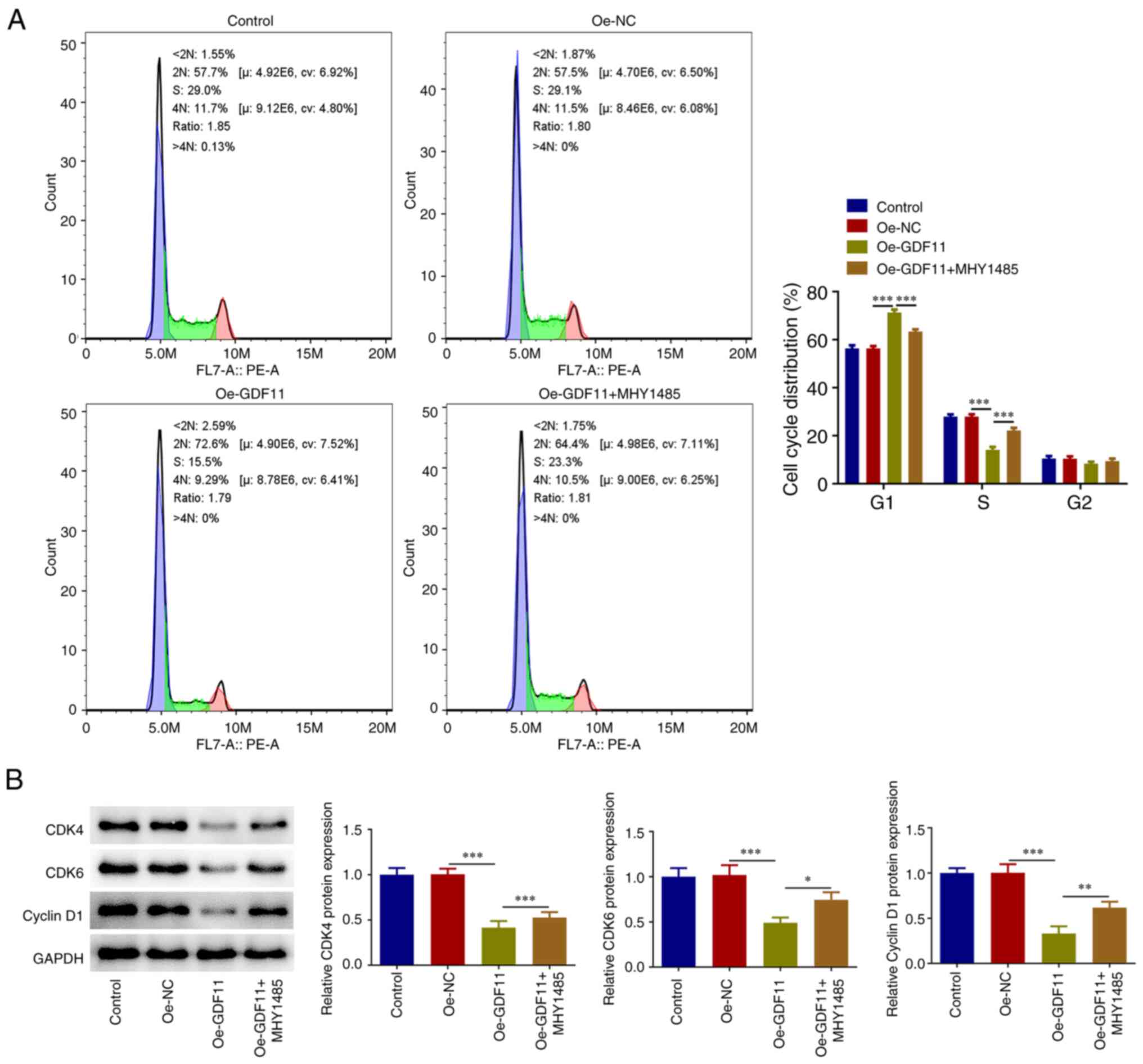
GDF11 induces HCC cell cycle arrest by
suppressing the mTORC1 signaling pathway
Flow cytometry was used for cell cycle distribution
analysis and it was revealed that GDF11 overexpression elevated the
proportion of cells at the G1 stage and reduced the
proportion of cells at the S stage compared with the Oe-NC group.
Furthermore, the results showed a decrease in the proportion of
cells at the G1 stage and an increase in the proportion
of cells at the S stage after MHY1485 treatment in comparison with
the Ov-GDF11 group (Fig. 5A).
GDF11 induced Huh-7 cell cycle arrest, which was partially reversed
by MHY1485 treatment. In addition, GDF11 overexpression reduced
CDK4, CDK6 and cyclin D1 protein expression levels compared with
those in the Oe-NC group, whereas MHY1485 treatment partially
reversed the regulatory effects of GDF11 overexpression on cell
cycle-associated proteins (Fig.
5B). Thus, GDF11 may induce HCC cell cycle arrest by
suppressing mTORC1 signaling pathway.
GDF11 inhibits HCC migration,
invasion, EMT and angiogenesis by suppressing mTORC1 signaling
pathway
As determined by wound healing and Transwell assays,
GDF11 overexpression suppressed the migration and invasion of Huh-7
cells compared with that in the Oe-NC group, which was partially
reversed by MHY1485 treatment (Fig.
6A and B). Tumors derive their
metastatic capacity through epithelial-mesenchymal transition (EMT)
(22). EMT-associated biomarkers
were thus detected. GDF11 overexpression elevated E-cadherin
protein expression levels, and reduced N-cadherin, Snail and
Vimentin protein expression levels compared with those in the Oe-NC
group, whereas MHY1485 treatment partially reversed the regulatory
effects of GDF11 overexpression on EMT-associated proteins
(Fig. 6C). These findings
indicated that GDF11 suppressed EMT in Huh-7 cells, which was
partially reversed by MHY1485 treatment. Angiogenesis is
responsible for nutritional provision of tumor metastasis (23). The results of the tube formation
assay revealed that GDF11 overexpression suppressed the in
vitro angiogenesis of HUVECs compared with the Oe-NC group,
whereas MHY1485 treatment partially reversed the suppressive effect
of GDF11 overexpression on angiogenic ability (Fig. 6D). Thus, GDF11 may inhibit HCC
metastasis by suppressing the mTORC1 signaling pathway.
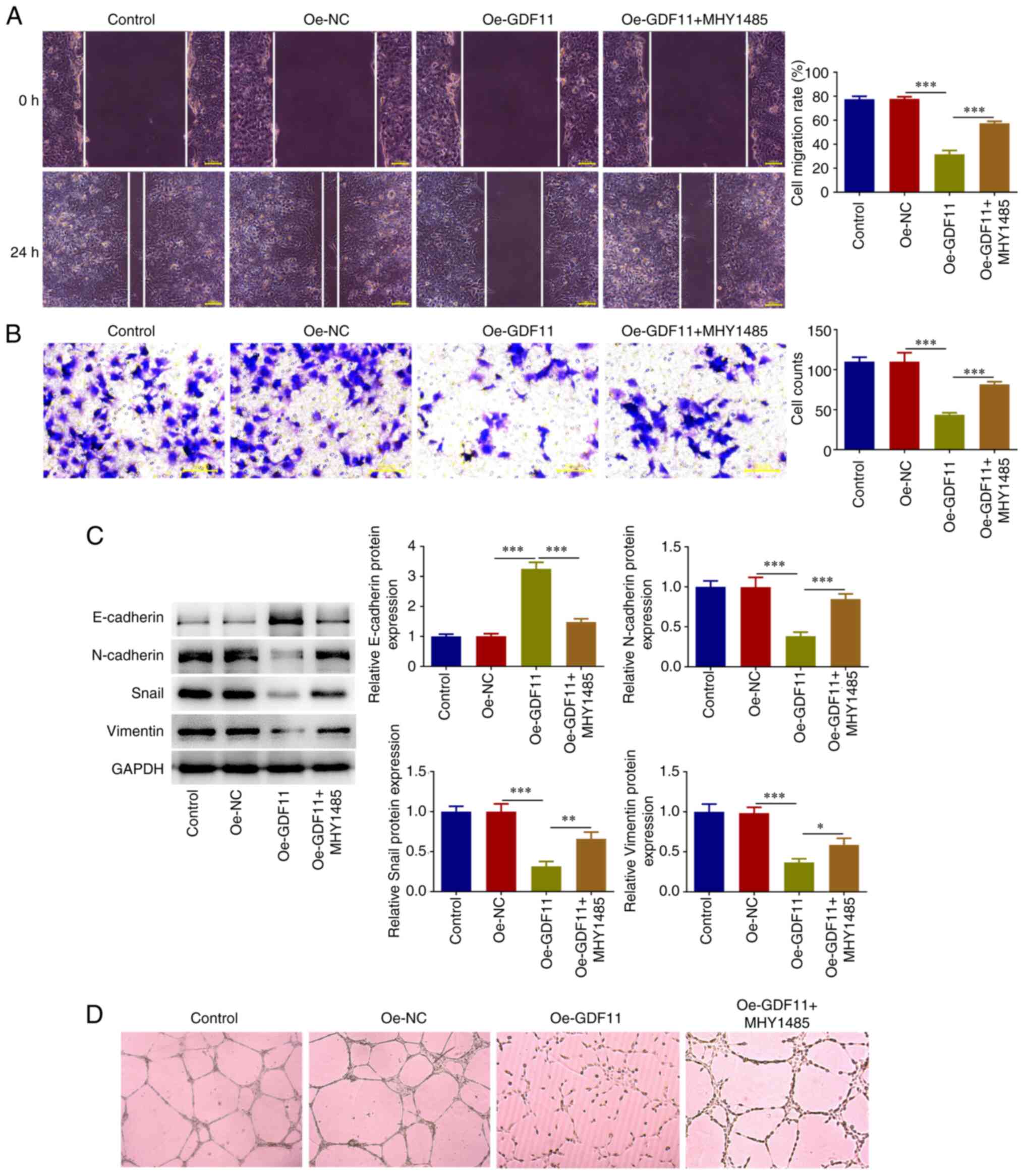 | Figure 6GDF11 inhibits hepatocellular
carcinoma migration, invasion, EMT and angiogenesis by suppressing
the mTORC1 signaling pathway. Huh-7 cells were transfected with
either Oe-GDF11 or Oe-NC. Oe-GDF11-transfected Huh-7 cells were
treated with the mTOR activator MHY1485. (A) Cell migration was
investigated using a wound healing assay. (B) Cell invasion was
investigated using a Transwell invasion assay. (C) E-cadherin,
N-cadherin, Snail and Vimentin protein expression levels were
detected by western blotting. (D) HUVECs were incubated with the
conditioned media of Huh-7 cells at 37˚C for 24 h. In vitro
angiogenesis of HUVECs was investigated using a tube formation
assay. *P<0.05, **P<0.01,
***P<0.001. GDF11, growth differentiation factor 11;
mTORC1, mammalian target of rapamycin complex 1; Oe, overexpression
plasmid; NC, negative control; HUVECs, human umbilical vein
endothelial cells. |
Discussion
As a major contributor to global cancer-related
mortality, HCC poses a challenging threat to public health
(24). The malignant
proliferation, migration and invasion of hepatoma cells triggers
the metastasis and recurrence of HCC, resulting in an unfavorable
prognosis of HCC (25,26). Therefore, the present study
investigated the functional role of GDF11 in HCC proliferation,
colony-forming ability, apoptosis, cell cycle progression,
migration, invasion, EMT and angiogenesis, aiming to provide novel
perspectives on the biological mechanisms of HCC and to identify
promising targets for HCC therapy.
In the present study, GDF11 was verified to be lowly
expressed in HCC cells. Overexpression of GDF11 inhibited the
proliferation, colony-forming ability, migration, invasion, EMT and
angiogenesis of HCC cells, and facilitated the apoptosis and cell
cycle arrest of HCC cells.
Dysregulated autophagy has been implicated in
various types of cancer, including HCC (27,28).
Luteolin can induce autophagy by increasing the number of
autophagosomes and enhancing Beclin-1 expression, thereby promoting
HCC cell apoptosis (29).
Knockdown of the oncogene UBE2I can inhibit cellular proliferation,
migration and invasion via the autophagy-related pathway in HCC
(30). The E2F1/USP11 positive
feedback loop can facilitate HCC cell proliferation and metastasis,
and can promote tumor growth in vivo, by activating the
ERK/mTOR pathway to inhibit autophagy (31). The novel mTOR inhibitor Torin-2 can
induce autophagy by inactivating mTORC1 to suppress HCC cell
proliferation and promote HCC cell apoptosis (32). In addition, DHX15 can inhibit
autophagy through the mTORC1 pathway, thereby promoting HCC cell
proliferation (33). In the
present study, it was verified that overexpression of GDF11
inactivated the mTORC1 signaling pathway to enhance autophagy in
HCC cells. MHY1485 is a potent cell-permeable mTOR activator, which
can induce the activation of mTOR via two possible mechanisms: i)
MHY1485 may bind a different site from an ATP-binding site of mTOR;
and ii) MHY1485 could indirectly activate mTOR through elevation of
p-mTOR at ser2448(34). Treatment
with the mTOR activator MHY1485 partially reversed the
tumor-suppressive effects of GDF11 overexpression on HCC in the
current study.
Literature reports that LAIR-1 can inhibit HCC cell
proliferation and invasion via suppressing the PI3K-AKT-mTOR
pathway (35). Furthermore, it has
been verified that GDF11 can regulate the PI3K-AKT pathway in HCC
cells (13). The results of the
current study indicated that GDF11 could inhibit proliferation and
colony formation, facilitate apoptosis, induce cell cycle arrest,
and restrain migration and invasion of HCC cells by suppressing the
mTORC1 signaling pathway. Therefore, AKT could be involved in the
mTOR-dependent mechanism of GDF11 action in HCC. The aforementioned
prospective molecular mechanisms require further investigation in
future studies.
In conclusion, GDF11 may exert tumor-suppressive
effects on HCC cells through inactivating the mTORC1 signaling
pathway to strengthen autophagy. These findings are beneficial to
the development of a promising approach for HCC therapy. Modulation
of GDF11 serves as an attractive marker for HCC prediction,
prevention and novel therapy. Notably, it has been verified that
spermidine can inhibit high glucose-induced endoplasmic reticulum
stress in HT22 cells via the upregulation of GDF11, and can prevent
liver fibrosis and HCC by activating MAP1S-mediated autophagy
(36,37). These previous findings indicated
that spermidine may be developed as a suitable drug candidate for
the induction of GDF11 in HCC treatment. In addition, the
exploration of more specific GDF11 agonists may be used to
upregulate GDF11 in HCC therapy. Furthermore, in vivo animal
experiments should be conducted in the future to further support
the obtained conclusions and to assess the predictive values of
GDF11 and the mTORC1-autophagy axis.
Acknowledgements
Not applicable.
Funding
Funding: The present study was funded by the Scientific Research
Fund of Hunan Provincial Education Department (grant no. 21C1386),
the Hunan Provincial Science and Technology Department (grant nos.
2021SK51204, 2021SK4047, 2022JJ50300 and 2021SK51201) and the
Huaihua Science and Technology Department (grant no.
2022R2203).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
QW contributed to study conception, designed the
research study, performed the experiments, collected the data,
performed data analysis, and wrote and critically revised the
manuscript. CF designed the research study, performed the
experiments, collected the data, performed data analysis and wrote
the manuscript. KL performed the experiments, collected data and
performed data analysis. JT contributed to study conception,
designed the research study, and wrote and critically revised the
manuscript. All authors read and approved the final manuscript, and
confirm the authenticity of all the raw data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Cao M, Ren Y, Li Y, Deng J, Su X, Tang Y,
Yuan F, Deng H, Yang G, He Z, et al: Lnc-ZEB2-19 inhibits the
progression and lenvatinib resistance of hepatocellular carcinoma
by attenuating the NF-κB signaling pathway through the TRA2A/RSPH14
axis. Int J Biol Sci. 19:3678–3693. 2023.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Kanwal F and Singal AG: Surveillance for
hepatocellular carcinoma: Current best practice and future
direction. Gastroenterology. 157:54–64. 2019.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Li CM, Zhang J, Wu W, Zhu Z, Li F, Wu D,
Wang XJ, Xie CM and Gong JP: FBXO43 increases CCND1 stability to
promote hepatocellular carcinoma cell proliferation and migration.
Front Oncol. 13(1138348)2023.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Liu Y, Yao Y, Liao B, Zhang H, Yang Z, Xia
P, Jiang X, Ma W, Wu X, Mei C, et al: A positive feedback loop of
CENPU/E2F6/E2F1 facilitates proliferation and metastasis via
ubiquitination of E2F6 in hepatocellular carcinoma. Int J Biol Sci.
18:4071–4087. 2022.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global cancer statistics 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249.
2021.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Yin Z, Dong C, Jiang K, Xu Z, Li R, Guo K,
Shao S and Wang L: Heterogeneity of cancer-associated fibroblasts
and roles in the progression, prognosis, and therapy of
hepatocellular carcinoma. J Hematol Oncol. 12(101)2019.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Walker RG, Poggioli T, Katsimpardi L,
Buchanan SM, Oh J, Wattrus S, Heidecker B, Fong YW, Rubin LL, Ganz
P, et al: Biochemistry and biology of GDF11 and myostatin:
Similarities, differences, and questions for future investigation.
Circ Res. 118:1125–1142. 2016.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Bajikar SS, Wang CC, Borten MA, Pereira
EJ, Atkins KA and Janes KA: Tumor-suppressor inactivation of GDF11
occurs by precursor sequestration in triple-negative breast cancer.
Dev Cell. 43:418–435.e13. 2017.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Gao H, He Z, Gao C, Liu N, Zhang Z, Niu W,
Niu J and Peng C: Exosome-transmitted miR-3124-5p promotes
cholangiocarcinoma development via targeting GDF11. Front Oncol.
12(936507)2022.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Ungaro F, Colombo P, Massimino L, Ugolini
GS, Correale C, Rasponi M, Garlatti V, Rubbino F, Tacconi C,
Spaggiari P, et al: Lymphatic endothelium contributes to colorectal
cancer growth via the soluble matrisome component GDF11. Int J
Cancer. 145:1913–1920. 2019.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Liu Y, Shao L, Chen K, Wang Z, Wang J,
Jing W and Hu M: GDF11 restrains tumor growth by promoting
apoptosis in pancreatic cancer. Onco Targets Ther. 11:8371–8379.
2018.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Frohlich J, Kovacovicova K, Raffaele M,
Virglova T, Cizkova E, Kucera J, Bienertova-Vasku J, Wabitsch M,
Peyrou M, Bonomini F, et al: GDF11 inhibits adipogenesis and
improves mature adipocytes metabolic function via WNT/β-catenin and
ALK5/SMAD2/3 pathways. Cell Prolif. 55(e13310)2022.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Frohlich J, Mazza T, Sobolewski C, Foti M
and Vinciguerra M: GDF11 rapidly increases lipid accumulation in
liver cancer cells through ALK5-dependent signaling. Biochim
Biophys Acta Mol Cell Biol Lipids. 1866(158920)2021.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Zhang YH, Pan LH, Pang Y, Yang JX, Lv MJ,
Liu F, Qu XF, Chen XX, Gong HJ, Liu D and Wei Y: GDF11/BMP11 as a
novel tumor marker for liver cancer. Exp Ther Med. 15:3495–3500.
2018.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Gerardo-Ramirez M, Lazzarini-Lechuga R,
Hernandez-Rizo S, Hernández-Rizo S, Jiménez-Salazar JE,
Simoni-Nieves A, García-Ruiz C, Fernández-Checa JC, Marquardt JU,
Coulouarn C, Gutiérrez-Ruiz MC, et al: GDF11 exhibits tumor
suppressive properties in hepatocellular carcinoma cells by
restricting clonal expansion and invasion. Biochim Biophys Acta Mol
Basis Dis. 1865:1540–1554. 2019.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Battaglioni S, Benjamin D, Wälchli M,
Maier T and Hall MN: mTOR substrate phosphorylation in growth
control. Cell. 185:1814–1836. 2022.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Han X, Goh KY, Lee WX, Choy SM and Tang
HW: The importance of mTORC1-autophagy axis for skeletal muscle
diseases. Int J Mol Sci. 24(297)2022.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Hernandez S, Simoni-Nieves A,
Gerardo-Ramirez M, Torres S, Fucho R, Gonzalez J, Castellanos-Tapia
L, Hernández-Pando R, Tejero-Barrera E, Bucio L, et al: GDF11
restricts aberrant lipogenesis and changes in mitochondrial
structure and function in human hepatocellular carcinoma cells. J
Cell Physiol. 236:4076–4090. 2021.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Saxton RA and Sabatini DM: mTOR signaling
in growth, metabolism, and disease. Cell. 168:960–976.
2017.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Chung CY, Shin HR, Berdan CA, Ford B, Ward
CC, Olzmann JA, Zoncu R and Nomura DK: Covalent targeting of the
vacuolar H+-ATPase activates autophagy via mTORC1
inhibition. Nat Chem Biol. 15:776–785. 2019.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Lamouille S, Xu J and Derynck R: Molecular
mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell
Biol. 15:178–196. 2014.PubMed/NCBI View
Article : Google Scholar
|
|
23
|
Wendong Y, Jiali J, Qiaomei F, Yayun W,
Xianze X, Zheng S and Wei H: Biomechanical forces and
force-triggered drug delivery in tumor neovascularization. Biomed
Pharmacother. 171(116117)2024.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Anwanwan D, Singh SK, Singh S, Saikam V
and Singh R: Challenges in liver cancer and possible treatment
approaches. Biochim Biophys Acta Rev Cancer.
1873(188314)2020.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Lee YS, Jeong S, Kim KY, Yoon JS, Kim S,
Yoon KS, Ha J, Kang I and Choe W: Honokiol inhibits hepatoma
carcinoma cell migration through downregulated Cyclophilin B
expression. Biochem Biophys Res Commun. 552:44–51. 2021.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Li T, Zhong J, Dong X, Xiu P, Wang F, Wei
H, Wang X, Xu Z, Liu F, Sun X and Li J: Meloxicam suppresses
hepatocellular carcinoma cell proliferation and migration by
targeting COX-2/PGE2-regulated activation of the β-catenin
signaling pathway. Oncol Rep. 35:3614–3622. 2016.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Mandell MA, Saha B and Thompson TA: The
tripartite nexus: Autophagy, cancer, and tripartite
motif-containing protein family members. Front Pharmacol.
11(308)2020.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Poillet-Perez L and White E: Role of tumor
and host autophagy in cancer metabolism. Genes Dev. 33:610–619.
2019.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Cao Z, Zhang H, Cai X, Fang W, Chai D, Wen
Y, Chen H, Chu F and Zhang Y: Luteolin promotes cell apoptosis by
inducing autophagy in hepatocellular carcinoma. Cell Physiol
Biochem. 43:1803–1812. 2017.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Wang XK, Liao XW, Zhou X, Han CY, Chen ZJ,
Yang CK, Huang JL, Wang JY, Liu JQ, Huang HS, et al: Oncogene UBE2I
enhances cellular invasion, migration and proliferation abilities
via autophagy-related pathway resulting in poor prognosis in
hepatocellular carcinoma. Am J Cancer Res. 10:4178–4197.
2020.PubMed/NCBI
|
|
31
|
Qiao L, Zhang Q, Sun Z, Liu Q, Wu Z, Hu W,
Bao S, Yang Q and Liu L: The E2F1/USP11 positive feedback loop
promotes hepatocellular carcinoma metastasis and inhibits autophagy
by activating ERK/mTOR pathway. Cancer Lett. 514:63–78.
2021.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Wang C, Wang X, Su Z, Fei H, Liu X and Pan
Q: The novel mTOR inhibitor Torin-2 induces autophagy and
downregulates the expression of UHRF1 to suppress hepatocarcinoma
cell growth. Oncol Rep. 34:1708–1716. 2015.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Zhao M, Ying L, Wang R, Yao J, Zhu L,
Zheng M, Chen Z and Yang Z: DHX15 inhibits autophagy and the
proliferation of hepatoma cells. Front Med (Lausanne).
7(591736)2021.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Choi YJ, Park YJ, Park JY, Jeong HO, Kim
DH, Ha YM, Kim JM, Song YM, Heo HS, Yu BP, et al: Inhibitory effect
of mTOR activator MHY1485 on autophagy: Suppression of lysosomal
fusion. PLoS One. 7(e43418)2012.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Zhou T, Liu L, Lan H and Fang D: Effects
of LAIR-1 on hepatocellular carcinoma cell proliferation and
invasion via PI3K-AKT-mTOR pathway regulation. Immun Inflamm Dis.
11(e982)2023.PubMed/NCBI View
Article : Google Scholar
|
|
36
|
Liao ZZ, Deng Q, Xiao F, Xie M and Tang
XQ: Spermidine inhibits high glucose-induced endoplasmic reticulum
stress in HT22 cells by upregulation of growth differentiation
factor 11. Neuroreport. 33:819–827. 2022.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Yue F, Li W, Zou J, Jiang X, Xu G, Huang H
and Liu L: Spermidine prolongs lifespan and prevents liver fibrosis
and hepatocellular carcinoma by activating MAP1S-mediated
autophagy. Cancer Res. 77:2938–2951. 2017.PubMed/NCBI View Article : Google Scholar
|