Introduction
Mixed gonadal dysgenesis (MGD) is a disorder of sex
development (DSD) characterized by mixed histological findings of
cord-like gonads, dysplastic testes, and asymmetrical internal and
external genitalia (1). This
condition has gained recognition as a common chromosomal
abnormality that occurs in 1/6,000 cases (2). The 45,X/46,XY mosaicism is a disorder
of sex development that has an estimated incidence of <1/15,000
live births. Each cell originates from a single fertilized egg and,
thereby, has the same genetic origin. However, the precise role of
gonadal differentiation in MGD remains elusive. Diverse proportions
of 45,X and 46,XY cells in the gonads can give rise to various
phenotypes-including bilateral testicles, unilateral testicles plus
other streak gonads, ovotestes, and bilateral streak gonads. This
proportion changes the total amount of testosterone secreted by the
bilateral gonads and determines the degree of masculinization of
the external genitalia (3,4). The frequency of gonadal tumor
complications in patients with MGD is 15-20%. Although the risk of
developing gonadoblastoma in individuals with 45,X/46,XY mosaicism
is approximately 15-30%, it varies with age (ranging between
approximately 3 and 4% at the age of 10 years, to approximately 46%
by age 40) (5). Gonadotropins,
particularly FSH, have tumor-promoting effects. Their levels are
elevated during puberty, resulting in an increased incidence of
tumors. Early prophylactic gonadectomy is recommended, given the
elevated risk of gonadoblastoma (an anaplastic germ cell tumor
[i.e., dysgerminoma]) or other high-grade tumors (3,5,6).
However, in diseases with 45,X/46,XY mosaicism, patients often have
problems with romantic relationships, owing to a lack of fertility
in their oocytes, in addition to abnormal secondary sexual
characteristics-as was also the case with our patient. We report a
case of MGD diagnosed as primary amenorrhea in a patient who
desired to have a child, wherein a gonadoblastoma was found after
prophylactic laparoscopic gonadectomy.
Case report
A 21-year-old patient with a female phenotype
presented to the hospital with primary amenorrhea, and expressed
her desire to have a child. The patient was unmarried and
nulligravida. Her height and weight were 146 cm and 67 kg (body
mass index, 31.3 kg/m2), respectively. Aside from
undergoing tympanoplasty for otitis media at the ages of 7, 9, 10,
and 11 years, the patient's medical history was unremarkable. The
patient's grandfather had lung cancer, and an uncle had esophageal
cancer. There was no family history of gynecological problems,
short stature, or consanguineous marriages.
The patient's was quite short in stature;
transabdominal ultrasonography revealed hypoplasia of the uterus,
which led to a suspicion of Turner's syndrome. The patient had
cubitus valgus and a webbed neck, wherein the external genitalia
were female. Transrectal ultrasonography (SONOVISTA GX30, Konica
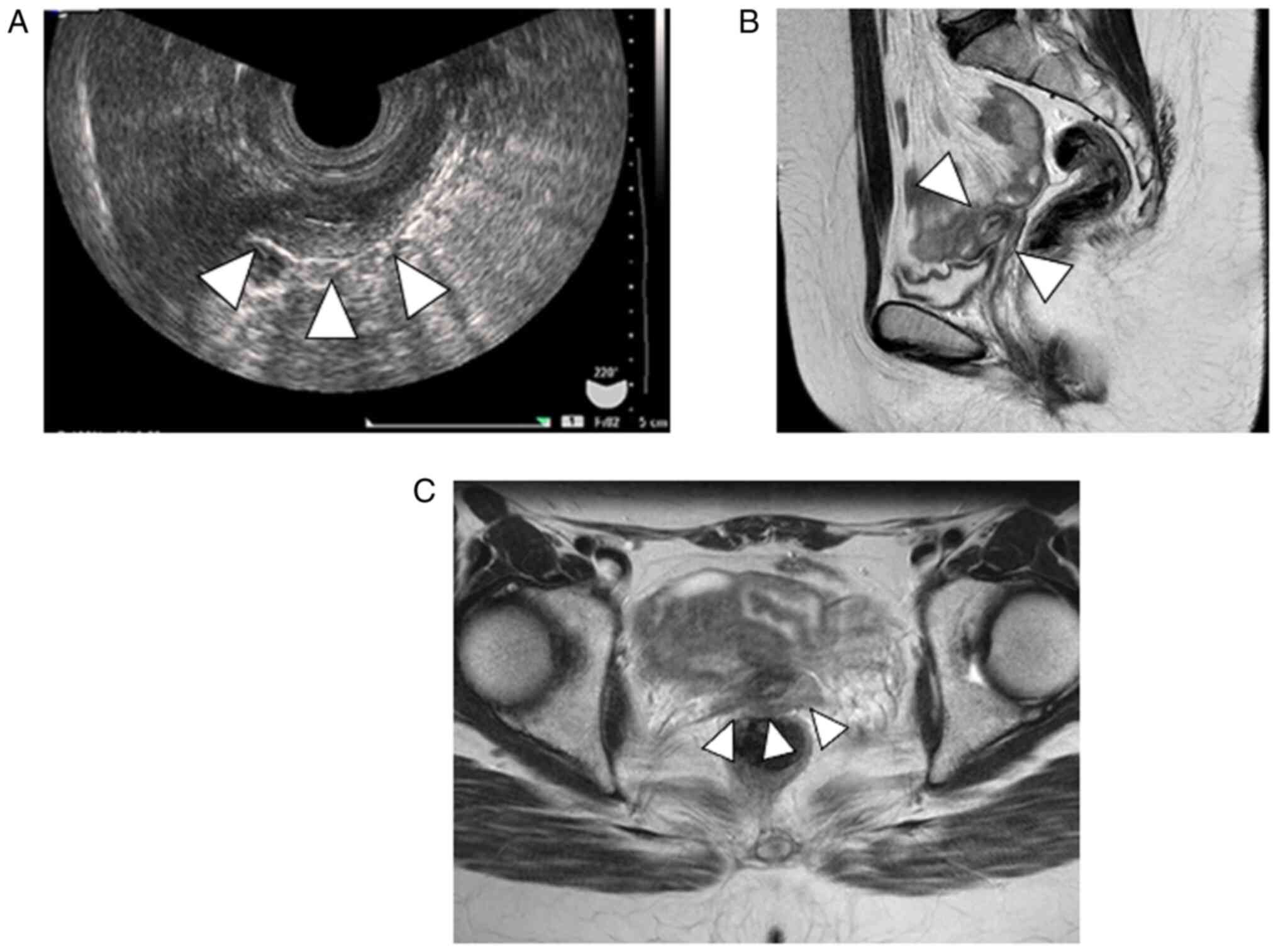
Minolta, 1 Sakura-machi, Hino-shi Tokyo 191-8511, Japan) revealed a
30 mm uterus and indistinct bilateral adnexa (Fig. 1A). Contrast-enhanced computed
tomography revealed no abnormalities in the cardiovascular system
or kidneys. Pelvic contrast-enhanced magnetic resonance imaging
(MRI) revealed a 30 mm-long uterus, vaginal thinning, and no
structures in the pelvis or inguinal region that could be
considered gonads (Fig. 1B and
C). Serum hormone tests revealed
the following levels: estradiol, ≤5.0 pg/ml (normal range: 13-70
pg/ml); luteinizing hormone, 25.5 mIU/ml (normal range: 1.8-7.0
mIU/ml); and follicle-stimulating hormone (FSH), 98.4 mIU/ml
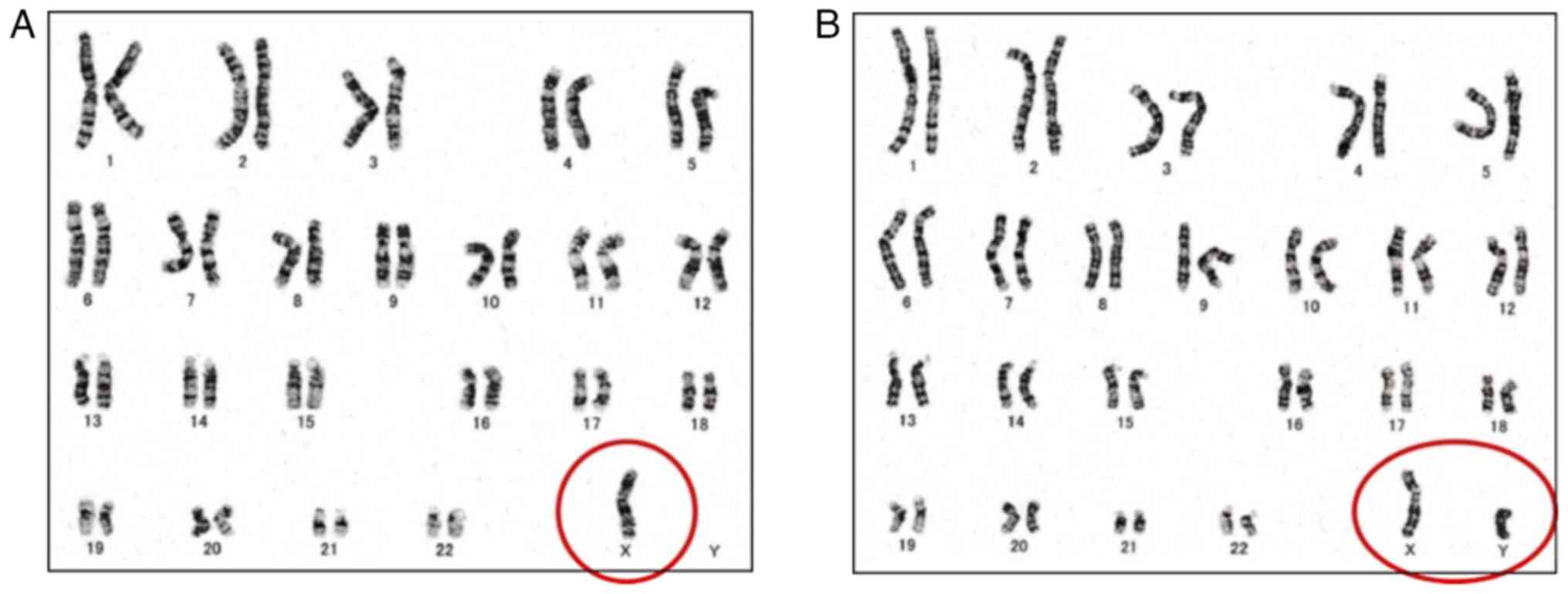
(normal range: 5.2-14.4 mIU/ml). Chromosomal testing via the G-band
method was performed of the patient's peripheral blood. The
resultant karyotype was 45,X [8]/46,XY [22] (45,X: 8 cells, 46,XY:
22 cells; Fig. 2). Chromosomal
testing via the G-band method was performed by SRL, Inc. Therefore,
the patient was diagnosed with 45,X/46,XY MGD. Kaufmann therapy was
initiated to induce menstruation and maintain her bone mass. Since
the patient wished to have a child, adequate counseling was
provided by our hospital's Department of Reproductive Medicine and
Genetic Medicine. The patient and her family, including her
partner, were informed of the difficulty in conceiving using
autologous oocytes, possibility of gonadal tumorigenesis, and her
elevated risk of developing a malignancy. After thorough counseling
for one year, consent was obtained for prophylactic gonadectomy.
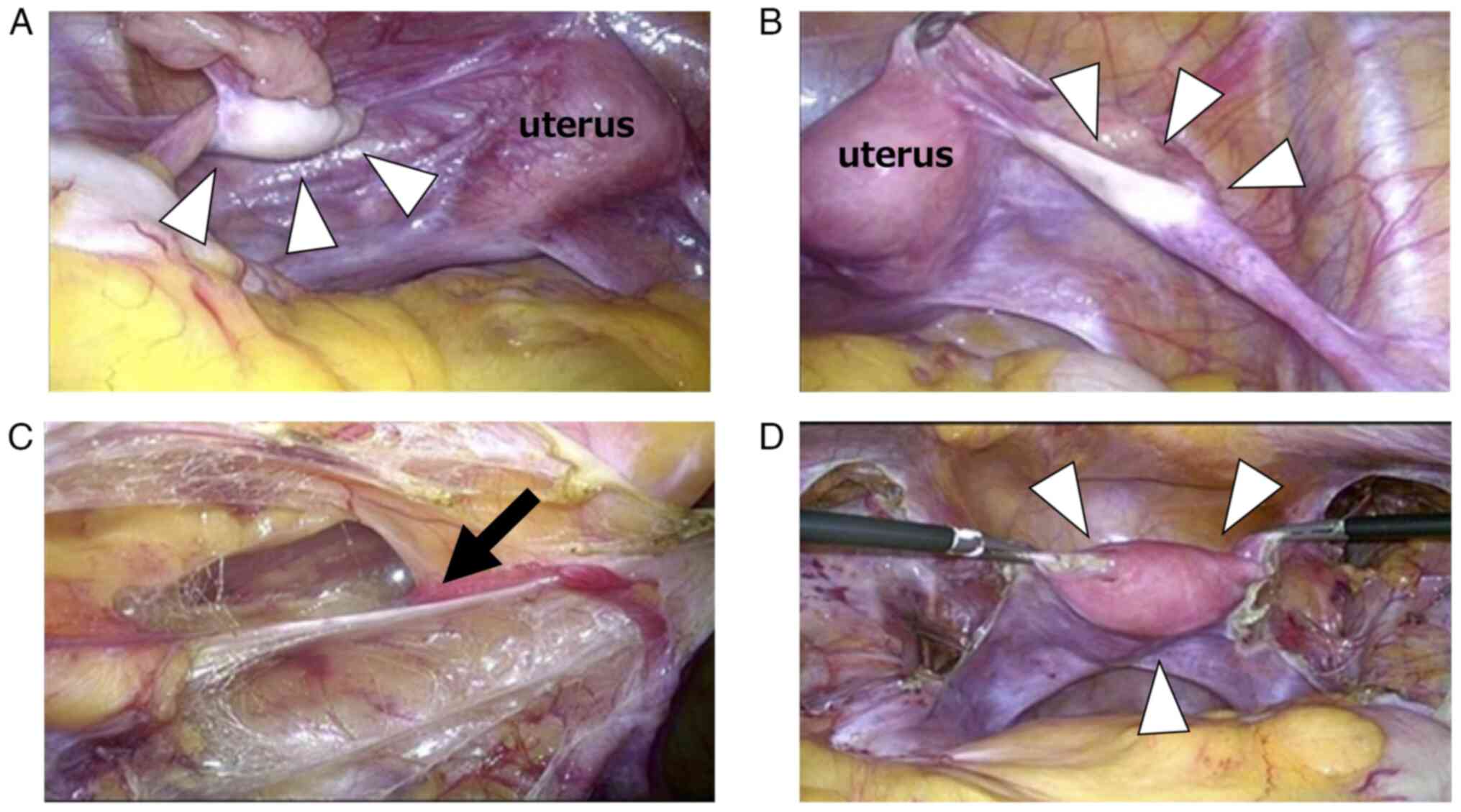
Imaging revealed no neoplastic lesions, and laparoscopic surgery
was performed. Surgical findings revealed a hypoplastic uterus with
a white structure and calcifications on the left side (1.5 cm), as
well as a cord-like structure on the right side (1.5 cm) that was
thought to be a gonad. The broad mesentery was incised along the
round ligament of the uterus toward the internal inguinal canal,
then expanded to the intersection of the round ligament of the
uterus and the inferior abdominal wall artery. Bilateral
gonadectomy was performed after confirming the absence of an
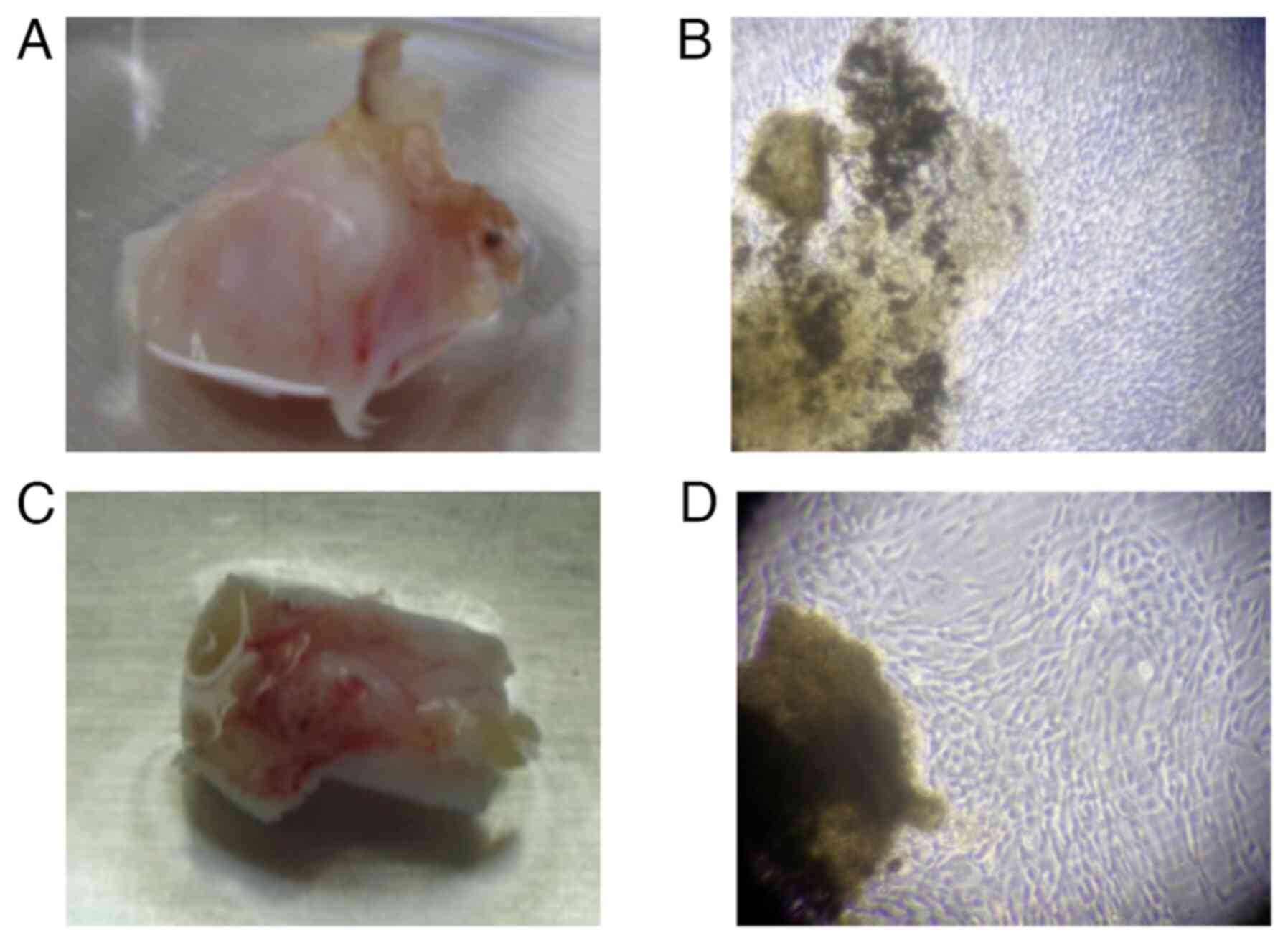
obvious mass (Fig. 3A-D). Ascites
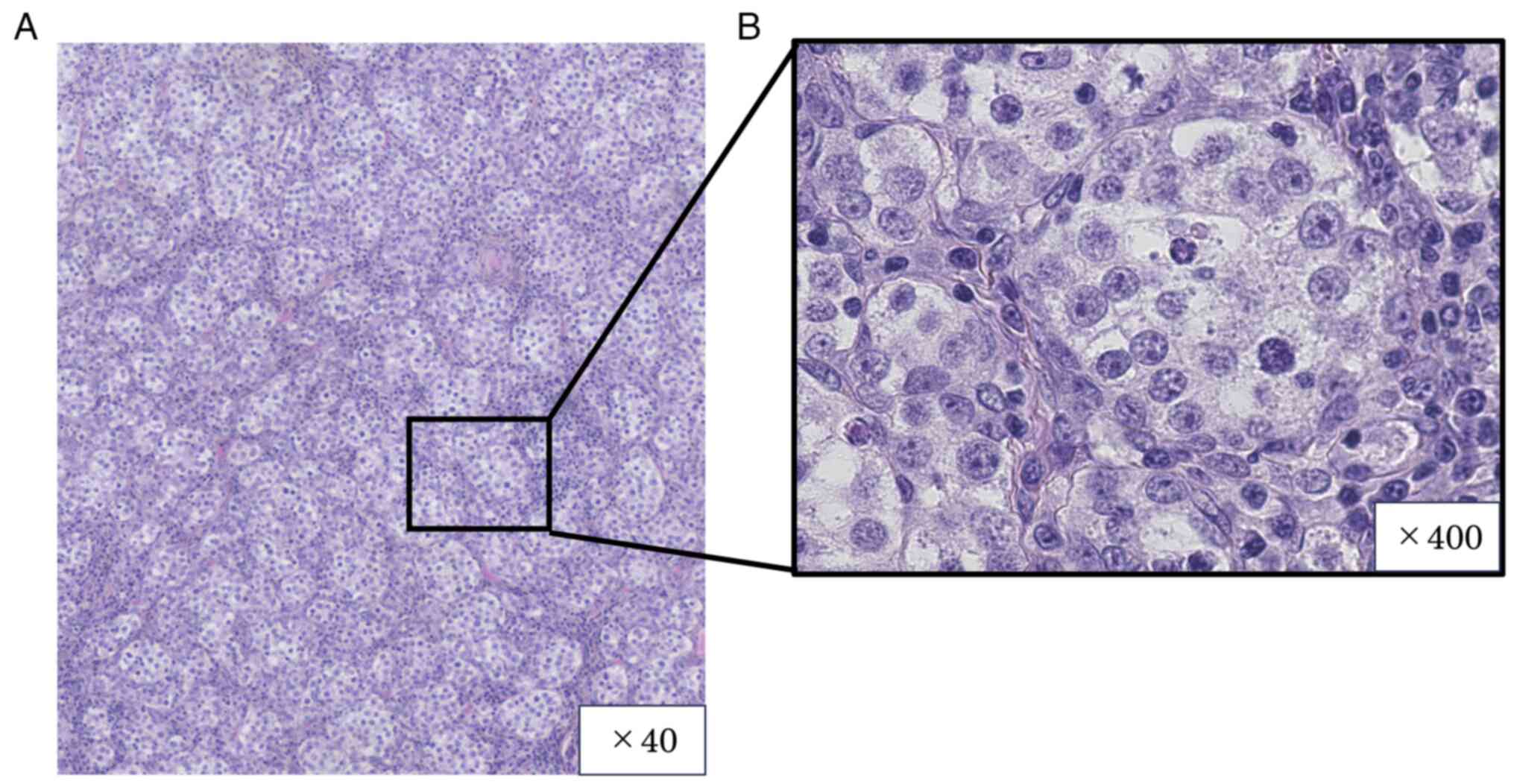
cytology results were negative. Histopathological examination of
the left gonadal tissue revealed immature tumor cells growing in
numerous small foci. The tumor cells exhibited pale sporangia,
nuclear atypia, and nuclear mitosis. Additionally, a small number
of large, weakly acidophilic, spore-associated tumor cells growing
within the tumor tissue were considered to be Sertoli cells
(Fig. 4A and B). All images of the hematoxylin- and
eosin-stained sections were obtained with an all-in-one
fluorescence microscope (BZ-X710, KEYENCE, Osaka, Japan). Normal
ovarian and fallopian tube tissues were observed in the right
gonad. The patient was diagnosed with stage IA (FIGO 2014) pT1aN0M0
gonadoblastoma. No additional postoperative systemic chemotherapy
was administered. The patient remained recurrence-free and
continuously received Kaufmann therapy, which included Estrana
Tapes (0.72 mg) (408 Tashirodaikan-machi, Tashiro, Tosu, Saga
841-0017, Japan) every 2 days and Provera (5 mg) (Pfizer, 66 Hudson
Boulevard East, New York, NY 10001-2192 USA) for 10 days during the
second half of the planned menstrual cycle, to induce proper
development of her uterus, as well as prevent cardiovascular
disorders and loss of bone density.
To evaluate the patient for gonadal mosaicism, cells
derived from the excised left and right gonads were cultured for 12
days (Fig. 5A-D). Chromosomal
testing (via the G-band method) was performed using 60 cells from
this primary culture. The resultant karyotypes were 45,X [23]/46,XY
[37] in the cells derived from the left gonad; and 45, X [38]/46,XY
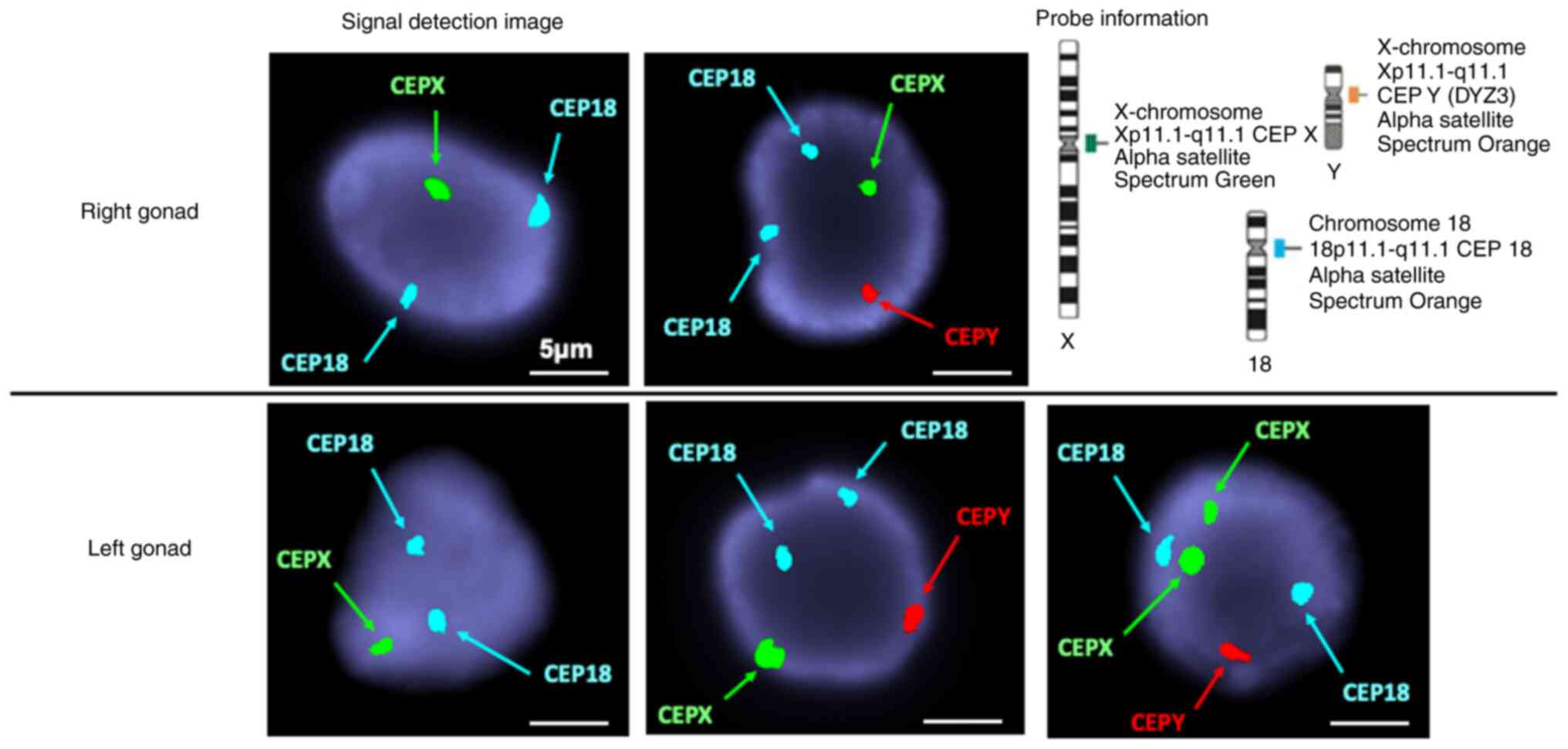
[22] in the cells derived from the right gonad (Table I). Fluorescence in situ
hybridization (FISH) was performed by OVUS Inc. FISH was performed
according to the manufacturer's protocol (Abbott Molecular Inc.,
1300 East Touhy Avenue Des Plaines, IL, USA) on uncultured tissue
fixed with Carnoy solution using the AneuVysion Multicolor DNA
Probe Kit (Vysis CEP 18/X/Y-alpha satellite/LSI 13/21). FISH was
performed on 100 uncultured cells derived from the left and right
gonads, using CEP-Y (Spectrum Orange), CEP-X (Spectrum Green), and
CEP-18 (Spectrum Aqua) probes. These probes hybridized to the
centromeric region of the Y chromosome (Yp11.1-q11.1), X chromosome
(Xp11.1-q11.1), and chromosome 18 (18p11.1-q11.1), respectively
(Fig. 6). The karyotypes were 45,X
[46]/46,XY [52]/47,XXY [2] in the cells derived from the left gonad
and 45,X [53]/46,XY [47] in the cells derived from the right gonad
(Table I).
 | Table IDistribution of karyotypes. |
Table I
Distribution of karyotypes.
| | Cells derived from
gonads |
|---|
| | G-band (60
cells) | FISH (100 uncultured
cells) |
|---|
| Gonad | X | XY | X | XY | XXY |
|---|
| Right | 38 (63%) | 22 (37%) | 53 | 47 | 0 |
| Left | 23 (38%) | 37 (62%) | 46 | 52 | 2 |
Discussion
MGD is one of the most prevalent chromosomal
abnormalities, characterized by 45,X/46,XY mosaicism. Initially
reported by Sohval in 1963(1). In
the present case, the patient was not diagnosed with MGD until her
20s, owing to experiencing primary amenorrhea. Primary amenorrhea
is caused by chromosomal abnormalities such as Turner's syndrome.
Other causes include hypothalamic, pituitary, or abnormal genital
tract differentiation. Chromosomal testing, using peripheral blood
lymphocytes, is often used for diagnosis. However, cultured cells
derived from the gonads more accurately reflect gonadal mosaic than
do peripheral blood lymphocytes. Therefore, performing chromosome
and FISH analyses using excised gonads is helpful. FISH analysis
can be used for detecting minute deletions and translocations
visually and reliably. Many cases have been reported in the
literature wherein a patient's peripheral blood lymphocyte
chromosomal composition proportion did not match their gonadal or
internal/external genital phenotypes. This is because the mosaic
ratio at the gonadal developmental stage determines the phenotype
of the gonads and subsequent internal and external genitalia.
However, the mosaic ratio may change over time, and chromosomal
testing results do not always reflect the clinical phenotype
(7). In the present case, the
percentage of 45,X cells was inconsistent with the results of
peripheral blood chromosomal testing (27%), whereas the percentages
in the left and right gonads were 38 and 63%, respectively. This
difference may have affected the total amount of testosterone
secreted, suggesting that the external genitalia were female.
Furthermore, FISH analysis of gonad-derived uncultured cells
revealed a slight XXY karyotype in the left gonad. Similarly to the
case in Turner's syndrome, the 45,X/46,XY mosaic may manifest short
stature, webbing of the neck, and external elbow caused by the 45,X
cells. Complications such as cardiac and vascular abnormalities,
abnormal kidney morphology, otitis media, hearing loss, obesity,
diabetes, hypothyroidism, and osteoporosis, may also occur
(8,9). Recurrent otitis media, short stature,
webbing of the neck and external elbow were observed in this case.
However, MGD had not been diagnosed at infancy because the
patient's external genitalia were female.
Patients with MGD are at high risk of developing
gonadal tumors, including gonadoblastoma (10). Gonadoblastoma is a tumor restricted
to the cordate gonads, with a Y chromosome kinetochore region
component (11). Gonadotropins,
particularly FSH, have tumor-promoting effects. Their levels are
elevated during puberty, resulting in an increased incidence of
tumors. Although gonadoblastomas are borderline tumors, they may
develop into highly malignant gonadal tumors such as dysgerminomas,
embryonal carcinomas, and mixed germ cell tumors (5,10).
Gonadoblastomas are present in 50% of patients with dysgerminoma
(10). Prophylactic gonadectomy
for Y chromosome-related sexually differentiated diseases should be
performed before puberty (5). The
prognosis of gonadoblastoma is favorable if the bilateral gonads,
including tumors, are excised-irrespective of the presence of
bilateral tumors. In the majority of cases involving pure
gonadoblastomas, no post-treatment is indicated. The present case
involved a 21-year-old woman with MGD who was at a high risk for
tumorigenesis. Gonadoblastoma is classified as a borderline
malignancy that should be carefully monitored.
Laparoscopic surgery is the preferred procedure for
prophylactic gonadectomy (12).
The gonads should be localized preoperatively if possible. However,
this is not always easy, because immature gonads may be ectopically
present. MRI is the most effective method for assessing the
abdominal cavity. However, if the location of the gonads cannot be
identified preoperatively, the course of the gonadal arteriovenous
vein can be used to identify the gonads. Gonads may also be present
in the inguinal canal in sexually differentiated diseases. In such
cases, ultrasonography may be used to locate the gonads. However,
if the gonads and tumors are not identified preoperatively,
adequate intraoperative observation may be necessary. In the
present case, a preoperative pelvic MRI did not reveal any
structures in the pelvis or inguinal region that could be
considered gonads. The presence of a mass in the inguinal canal was
confirmed via laparoscopic surgery; masses that could be considered
gonads were removed from both sides of the uterus, based on the
course of the gonadal arteriovenous vein.
Notification and genetic counseling in cases of
chromosomal abnormalities is a comprehensive process that includes:
(i) diagnosis and notification; (ii) advice regarding treatment,
health care, recuperation, and welfare; and (iii) family planning
counseling. During these processes, consideration of the
psychological burden on patients and their families is necessary
(12). Gonadectomy is recommended
due to the risk of developing gonadal tumors. However,
understanding and accepting this disease can be difficult. Building
relationships with patients and their families through repeated
counseling sessions is essential. For appropriate treatment and
management, implementing a team-based approach with collaboration
between multiple professionals is important, such as genetic and
reproductive medicine departments, in addition to the patients and
their families.
The 45X/46XY karyotype is characterized by one X
chromosome. As a result, any primordial follicles that form are
quickly depleted. In such cases, markedly elevated FSH levels
indicate a near absence of primordial follicles in the gonads,
rendering achieving pregnancy with autologous oocytes extremely
challenging. Nonetheless, there have been reports in the literature
from outside Japan describing the preservation of primordial
follicles following gonad removal. Although no reports have yet
confirmed successful pregnancies resulting from this preservation
method, future reports on the subject are warranted (13). Alternatively, pregnancy can be
achieved through egg donation (this is not currently feasible in
Japan) or in vitro fertilization. Some cases wherein
pregnancy was achieved with uterine preservation have also been
reported (14).
This case report has some limitations. First, it
describes only one case, while this disease is known to present in
diverse forms. Additionally, whether uterine preservation is
recommended and whether it is necessarily advisable to perfom a
gonadectomy as soon as possible remain unknown due to the short
tracking period. Nevertheless, 45X,46XY MGD is a rare disease and
this report aims to draw attention to this rare condition.
In conclusion, we report a case of MGD diagnosed
after primary amenorrhea and prophylactic gonadectomy, in which a
gonadoblastoma was detected. Early diagnosis of MGD is important,
as the risk of developing a highly malignant tumor in the gonads
increases after puberty. In cases of primary amenorrhea, this
disease should be considered in the differential diagnosis, for
which prophylactic gonadectomy is recommended as early as possible
after diagnosis. Management should also involve sufficient
counseling by a multidisciplinary team to help relieve the
psychological burden of the disease.
Acknowledgements
The authors would like to thank Dr Yuki Naru (OVUS
Inc. Nagoya, Aichi, Japan) for his cooperation with the chromosome
testing.
Funding
Funding: No funding was received.
Availability of data and materials
The data generated in this study are included in the
figures and/or tables of this article.
Authors' contributions
TU was a major contributor in writing the manuscript
under the guidance of IK. TU and IK confirm the authenticity of all
the raw data. TU, IK, KN and HK contributed to analysis and
interpretation of the data, and assisted in the preparation of the
manuscript. TU, IK, TK, AM, RO, AN, YE, YS, KN, HM, YO, YT, SN, KT,
MS, TY, YM, KB and YK have contributed to data collection and
interpretation, and critically reviewed the manuscript.
All authors read and approved the final version of
the manuscript, and agree to be accountable for all aspects of the
work in ensuring that questions related to the accuracy or
integrity of any part of the work are appropriately investigated
and resolved.
Ethics approval and consent to
participate
Our hospital's guidelines exempt case reports from
the need for ethics review.
Patient consent for publication
Written informed consent was obtained from the
patient to publish this case report.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Sohval AR: Hermaphroditism with ‘atypical’
or ‘mixed’ gonadal dysgenesis. Relationship To Gonadal Neoplasm. Am
J Med. 36:281–292. 1964.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Chang HJ, Clark RD and Bachman H: The
phenotype of 45,X/46,XY mosaicism: An analysis of 92 prenatally
diagnosed cases. Am J Hum Genet. 46:156–167. 1990.PubMed/NCBI
|
|
3
|
Huang YC, Lee CT, Wu MZ, Liu SY, Tung YC,
Ho HN and Tsai WY: The spectrum of 45,X/46,XY mosaicism in
Taiwanese children: The experience of a single center. J Formos Med
Assoc. 118 (1 Pt 3):450–456. 2019.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Rosa RF, D'Ecclesiis WF, Dibbi RP, Rosa
RC, Trevisan P, Graziadio C, Paskulin GA and Zen PR: 45,X/46,XY
mosaicism: Report on 14 patients from a Brazilian hospital. A
retrospective study. Sao Paulo Med J. 132:332–338. 2014.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Kriplani A, Agarwal N, Parul Sharma MC and
Manchanda R: Bilateral seminomas in a 45X/46XY mosaic with Turner's
phenotype: An unusual case of mixed gonadal dysgenesis. J Obstet
Gynaecol Res. 29:63–66. 2003.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Lee PA, Houk CP, Ahmed SF and Hughes IA:
International Consensus Conference on Intersex organized by the
Lawson Wilkins Pediatric Endocrine Society and the European Society
for Paediatric Endocrinology. Consensus statement on management of
intersex disorders. International consensus conference on intersex.
Pediatrics. 118:e488–e500. 2006.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Takahashi I, Miyamoto J and Hasegawa Y:
Limitations of G-banding karyotype analysis with peripheral
lymphocytes in diagnosing mixed gonadal dysgenesis. Clin Pediatr
Endocrinol. 15:109–115. 2006.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Farrugia MK, Sebire NJ, Achermann JC,
Eisawi A, Duffy PG and Mushtaq I: Clinical and gonadal features and
early surgical management of 45,X/46,XY and 45,X/47,XYY chromosomal
mosaicism presenting with genital anomalies. J Pediatr Urol.
9:139–144. 2013.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Berberoğlu M and Şıklar Z: Ankara
University Dsd Ethic Committee. The evaluation of cases with
Y-chromosome gonadal dysgenesis: Clinical experience over 18 years.
J Clin Res Pediatr Endocrinol. 10:30–37. 2018.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Verp MS and Simpson JL: Abnormal sexual
differentiation and neoplasia. Cancer Genet Cytogenet. 25:191–218.
1987.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Page DC: Hypothesis: A Y-chromosomal gene
causes gonadoblastoma in dysgenetic gonads. Development. 101
Suppl:S151–S155. 1987.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Weidler EM, Pearson M, van Leeuwen K and
Garvey E: Clinical management in mixed gonadal dysgenesis with
chromosomal mosaicism: Considerations in newborns and adolescents.
Semin Pediatr Surg. 28(150841)2019.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Siebert AL, Gomez-Lobo V, Johnson EK,
Nahata L, Orwig KE, Pyle LC, Witchel SF, Finlayson C and Laronda
MM: Differences in gonadal tissue cryopreservation practices for
differences of sex development across regions in the United States.
Front Endocrinol (Lausanne). 13(990359)2023.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Urban A, Knap-Wielgus W, Grymowicz M and
Smolarczyk R: Two successful pregnancies after in vitro
fertilisation with oocyte donation in a patient with Swyer
syndrome-a case report. Prz Menopauzalny. 20:158–161.
2021.PubMed/NCBI View Article : Google Scholar
|