Introduction
Venous thromboembolism, including deep vein
thrombosis (DVT), and its severe form pulmonary embolism (PE), is
the third most frequent complication of cardiovascular diseases,
affecting ~400,000 people annually (1,2).
Currently, compression ultrasound is the first line imaging
modality for the diagnosis of DVT (3). The treatment of DVT consists of
surgical invention, such as thrombectomy or catheter-based
thrombolysis and drug prevention including anticoagulation by
heparin, thrombin and vitamin K antagonists (4,5).
However, due to the low specificity (40-50%) of the diagnosis and
the limited efficacy of these treatments, patients at the acute
stage of DVT may develop PE, or even post-thrombotic syndrome,
which is the most common long-term complication in patients with
DVT, seriously threatening their survival and quality of life
(6-8).
Therefore, the progress of novel therapeutic strategies can be
achieved by improving the understanding of the pathophysiology of
DVT.
Previous evidence confirmed that oxidative stress
and inflammation are common pathological processes responsible for
vascular endothelial cell damage, which is one of the most
important causes leading to DVT (9-11).
Advanced glycation end products (AGEs) are heterogeneous molecules
produced by the non-enzymatic glycation of proteins and lipids
under hyperglycemic or oxidative stress conditions (12). These molecules can bind with their
receptor [receptor of advanced glycation end products (RAGE)] to
evoke an inflammatory response and induce oxidative stress, as well
as thrombogenic reactions, playing a central role in the
development of vascular complications (11,13-15).
The previous study conducted by the authors demonstrated the
involvement of AGEs/RAGE in the development of human umbilical vein
endothelial cell (HUVEC) injury. The RAGE inhibitor downregulated
endothenin-1 (ET-1) levels, which is considered to be the hallmark
of endothelial injury, thereby mitigating HUVEC injury (16). Therefore, blockade of AGEs/RAGE is
an effective approach for the prevention of DVT.
Peroxisome proliferator-activated receptor γ
(PPARγ), a member of the PPAR family of highly conserved nuclear
hormone receptors, is widely known for its important role in
regulating adipocyte differentiation, blood pressure, lipid
metabolism and insulin sensitivity (17). A previous study has reported the
eliminating effect of the activation of PPARγ on
hypercoagulability, one of the major factors contributing to DVT,
by inhibiting thrombin-induced platelet aggregation, which
indicates a potential role of PPARγ during the process of DVT
(18). It was also reported that
the activation of PPARγ may prevent thrombosis by downregulating
the expression of pro-inflammatory cell adhesion molecules and by
enhancing endothelial nitric oxide production. Of note, PPARγ has
been revealed to be associated with the AGE/RAGE axis to exert its
protective role against vascular inflammation and oxidative stress
(19). Chrysin, a natural
flavonoid, which acts as a PPARγ agonist, has shown the ability to
reduce serum AGE levels, inhibit AGEs-RAGE-mediated oxidative
stress and inflammation and attenuate endothelial dysfunction
(20). Previous studies have
confirmed the protection of PPARγ on endothelial cells (21-23);
however, whether PPARγ participates into AGE-RAGE-triggered
endothelial dysfunction, including oxidative stress and
inflammation, during DVT, remains poorly elucidated. Furthermore,
AGE-RAGE signaling is a pivotal inducer of endoplasmic reticulum
stress (ERS), which is closely associated with inflammation and
oxidative stress (24). PPARγ has
also been shown to be a critical mediator of ERS, as the inhibition
of it was demonstrated to relieve ERS and reduce the production of
pro-inflammatory cytokines and reactive oxygen species (ROS),
thereby alleviating ischemia-reperfusion injury (25).
The present study not only investigated the role of
PPARγ on AGEs-RAGE-triggered HUVEC injury, but also explored the
potential mechanism of action of PPARγ. The findings of the present
study contributed to the understanding of the pathophysiology of
DVT and offered novel strategies for the prevention of DVT.
Materials and methods
Cell culture and induction
HUVECs (cat. no. iCell-h110) were purchased from
iCell Bioscience, Inc. and were incubated in Endothelial Cell
Culture Medium (Cellverse Bioscience Technology Co., Ltd.)
containing 100 U/ml penicillin and 100 µg/ml streptomycin in the
presence of 5% CO2 at 37˚C. To mimic AGE-induced DVT,
HUVECs were treated with modified glycated human serum albumin
(M-HSA) for 24 h (26) and M-HSA
was prepared by co-incubation of HSA (15 mg/ml; Sigma-Aldrich;
Merck KGaA) and 3-deoxyglucosone (3-DG, 1 mM; Sigma-Aldrich; Merck
KGaA) for 2 weeks as described in a previous study conducted by the
authors (16). Tunicamycin (TM;
Sigma-Aldrich; Merck KGaA), the agonist of ERS, was used for
pre-treatment (5 µg/ml) for 6 h prior to M-HSA induction in
HUVECs.
Cell transfection
The sequences of PPARγ were cloned into the pcDNA
3.1 vector (Invitrogen; Thermo Fisher Scientific, Inc.) to
establish a PPARγ overexpression vector (oe-PPARγ). The pcDNA 3.1
vector was used as a negative control (oe-NC). HUVECs were
transfected with oe-PPARγ (15 nM) or oe-NC (15 nM) using
Lipofectamine™ 3000 reagent (Thermo Fisher Scientific, Inc.) at
37˚C strictly in line with the manufacturer's guidelines upon
reaching 80% confluence. At 48 h post-transfection, the transfected
HUVECs were harvested for subsequent experiments.
PPARγ activity assay
PPARγ activity was evaluated as previously reported
(27). In brief, nuclear extracts
were initially obtained from cultured HUVECs using a Nuclear
Extraction Kit (cat. no. ab113474; Abcam). Subsequently, PPARγ
activity was determined using a PPARγ Transcription Factor Assay
Kit (cat. no. ab133101; Abcam) by measuring the absorbance at 450
nm.
Cell viability assay
Cell viability was determined using the Cell
Counting Kit-8 (CCK-8) assay (16). In brief, HUVECs were cultured in
96-well plates (3.0x103 cells/well) and incubated for
24, 48 and 72 h, respectively. At different time points, 10 µl
CCK-8 solution (cat. no. KGA9305-500; Nanjing KeyGen Biotech Co.,
Ltd.) was added into each well and HUVECs were cultured in the
incubator for an additional 2 h at 37˚C. Finally, the absorbance at
450 nm of each well was detected using a microplate reader.
Terminal
deoxynucleotidyl-transferase-mediated dUTP nick end labeling
(TUNEL) assay
Cell apoptosis was assessed using the TUNEL assay
(28). HUVECs (5x104
cells/well) were cultured in 6-well plates with cell culture
silicon slides. Following treatment, the cells were fixed with 4%
paraformaldehyde for 30 min, permeabilized with 0.2% Triton X-100
for 5 min and blocked with 3% BSA (Wuhan Servicebio Technology Co.,
Ltd.) for 30 min at room temperature. Subsequently, the cells were
incubated with a TUNEL reaction mixture (Roche Diagnostics) at 37˚C
for 1 h and the cell nuclei were stained with 1 mg/ml
4',6-diamidino-2-phenylindole (Invitrogen, Thermo Fisher
Scientific, Inc.) for 10 min at 37˚C in the dark. Anti-fluorescence
quenching liquid was used for sealing. The apoptotic cells were
observed in five random fields using an inverted fluorescence
microscope (Olympus IX71; Olympus Corporation).
ELISA
The culture medium was harvested and centrifugated
at 12,000 x g, 4˚C for 10 min and the supernatant was then
collected. ELISA kits for 6-keto prostaglandin-F1 α (6-K-PGF1α;
E-EL-0054, Elabscience), ET-1 (ml025101), TNF-α (ml077385), IL-1β
(ml058059) and IL-6 (ml028583; all from Shanghai Enzyme-linked
Biotechnology Co., Ltd.) were applied to evaluate the corresponding
protein levels in the culture supernatant in accordance with the
manufacturer's instructions (16).
Assessment of oxidative stress
The levels of ROS, malondialdehyde (MDA) and
superoxide dismutase (SOD) were detected to evaluate the degree of
oxidative stress (16). For ROS
measurement, HUVECs were stained with 20 µM
2',7'-dichlorodihydrofluorescein diacetate strictly in line with
the instructions of the Fluorometric Intracellular ROS Kit (cat.
no. MAK143; Sigma-Aldrich; Merck KGaA). The fluorescence intensity
was detected by a microplate fluorometer (Molecular Devices, LLC).
For MDA and SOD measurements, the cell supernatant was measured
with Lipid Peroxidation MDA Assay Kit (cat. no. S0131S) and Total
Superoxide Dismutase Assay Kit (cat. no. S0101S) (both from
Beyotime Institute of Biotechnology) in accordance with the
manufacturer's guidelines, respectively.
Western blot analysis
Total protein was extracted from cells using a
radioimmunoprecipitation assay lysis buffer (Beyotime Institute of
Biotechnology), followed by the determination of protein
concentrations using an Enhanced BCA protein assay kit (Beyotime
Institute of Biotechnology). The same amount (30 µg/lane) of
protein was separated by 12% sodium dodecyl sulfate-polyacrylamide
gel electrophoresis and transferred onto polyvinylidene fluoride
membranes. Following blocking with 5% non-fat milk at room
temperature for 2 h, the membranes were incubated with primary
antibodies against PPARγ (1:1,000; cat. no. ab178860; Abcam), C/EBP
homologous protein (CHOP; 1:1,000; cat. no. 2895; Cell Signaling
Technology, Inc.), glucose-regulated protein 78 (GRP78; 1:1,000;
cat. no. ab21685; Abcam), phosphorylated (p)-protein kinase
(PKR)-like ER kinase (p-PERK; 1:200; cat. no. orb504147; Biorbyt),
PERK (1:500; cat. no. orb1294328; Biorbyt), p-inositol requiring
enzyme 1α (p-IRE1α; 1:1,000; cat. no. ab243665; Abcam), IRE1α
(1:1,000; cat. no. ab37073; Abcam) and GAPDH (1:2,500; cat. no.
ab9485; Abcam) at 4˚C overnight. On the following day, the
membranes were washed with Tris-buffered saline containing 0.1%
Tween-20, and subsequently incubated with horseradish
peroxidase-conjugated goat anti-rabbit (1:5,000; cat. no. ab6721;
Abcam) or goat anti-mouse (1:5,000; cat. no. ab6789; Abcam)
secondary antibodies at room temperature for 2 h. The signals were
visualized using Amersham ECL Prime Western Blotting Detection
Reagent (Amersham; Cytiva) and were semi-quantified by ImageJ
software (Version 1.52; National Institutes of Health).
Statistical analysis
All data were expressed as mean ± standard deviation
from at least three independent experiments. Data analysis was
conducted using GraphPad Prism 8 (GraphPad Software; Dotmatics).
One-way ANOVA analysis followed by the Tukey's post-hoc test was
used to compare the differences among groups. P<0.05 was
considered to indicate a statistically significant difference.
Results
PPARγ restores cell viability loss,
apoptosis and the levels of 6-K-PGF1α and ET-1 in M-HSA-stimulated
HUVECs
To investigate the role of PPARγ in AGE-induced VT,
HUVECs were stimulated by M-HSA to mimic AGE-induced DVT and the
expression levels of PPARγ were detected. As demonstrated in
Fig. 1A, the protein expression
levels of PPARγ were significantly reduced following M-HSA
stimulation in HUVECs. Therefore, a gain-of function experiment was
conducted to upregulate PPARγ (Fig.
S1). The expression levels of PPARγ in the M-HSA + oe-PPARγ
group were significantly higher than those in the M-HSA + oe-NC
group (Fig. 1A). In addition,
PPARγ activity was also weakened by M-HSA stimulation while it was
increased following PPARγ overexpression (Fig. 1B). Subsequently, the data from the
CCK-8 and TUNEL assays indicated that M-HSA led to a significant
reduction in cell viability and an apparent elevation in
TUNEL-positive cells, whereas these changes were inhibited when
PPARγ was overexpressed (Fig.
1C-E), suggesting that PPARγ had the ability to alleviate
M-HSA-induced cell viability loss and apoptosis in HUVECs. In
addition, the downregulated 6-K-PGF1α levels and upregulated ET-1
levels in HUVECs, which were caused following M-HSA induction, were
also partly abolished by PPARγ overexpression (Fig. 1F and G). These data suggested that PPARγ
overexpression attenuated M-HSA-induced endothelial injury in
HUVECs by improving cell viability, inhibiting cell apoptosis,
upregulating 6-K-PGF1α levels and downregulating ET-1 levels.
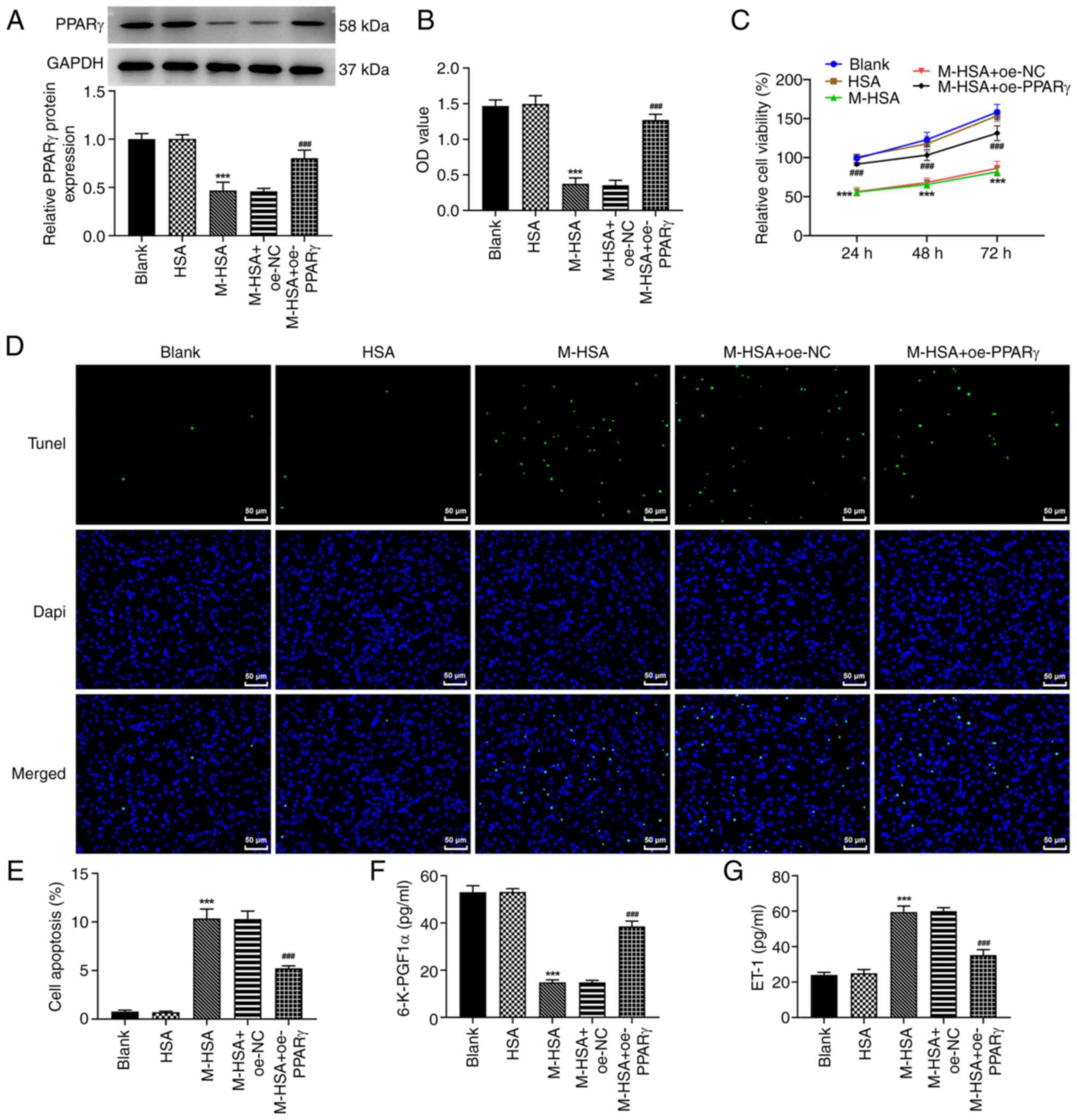 | Figure 1PPARγ restores cell viability loss,
apoptosis and levels of 6-K-PGF1α and ET-1 in M-HSA-stimulated
HUVECs. HUVECs were stimulated by M-HSA for 24 h to mimic advanced
glycation end products-induced vein thrombosis. Meanwhile, HUVECs
were transfected with oe-PPARγ or oe-NC for 48 h. (A) The protein
expression level of PPARγ was detected using western blot. (B) The
PPARγ activity was assessed at the absorbance of 450 nm. (C) Cell
viability was evaluated using Cell Counting Kit-8 assay at
indicated time points (24, 48 and 72 h). (D and E) Cell apoptosis
was determined using TUNEL assay. The concentrations of (F)
6-K-PGF1α and (G) ET-1 were measured by ELISA.
***P<0.001 vs. HSA and ###P<0.001 vs.
M-HSA + oe-NC. PPARγ, proliferator-activated receptor γ; 6-K-PGF1a,
6-keto prostaglandin-F1α; ET-1, endothenin-1; M-HSA, modified
glycated human serum albumin; HUVECs, human umbilical vein
endothelial cells; oe, overexpressing; NC, negative control. |
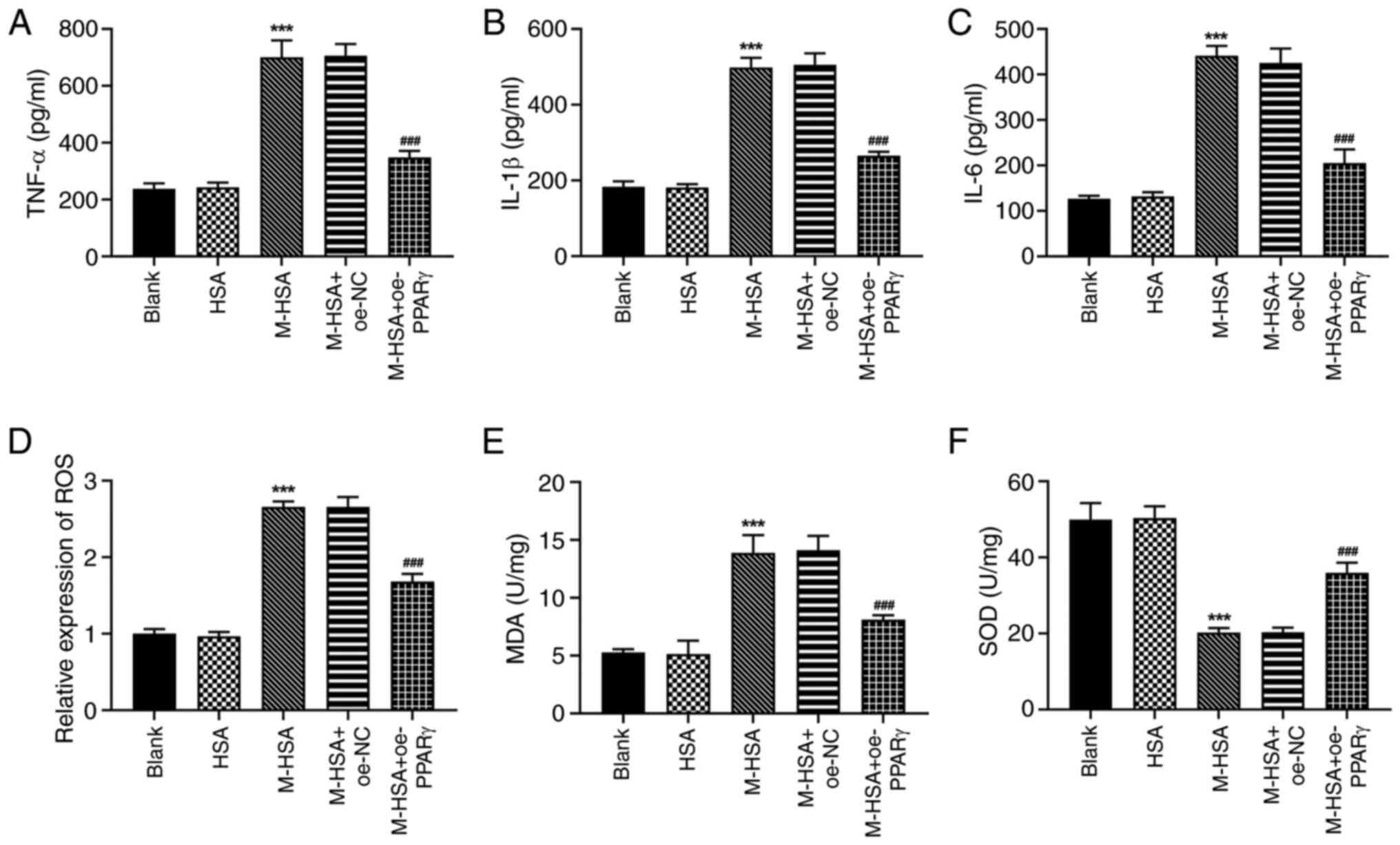
PPARγ reduces the induction of
inflammation and oxidative stress in M-HSA-stimulated HUVECs
Since oxidative stress and inflammation are common
pathological processes responsible for vascular endothelial cell
damage, the regulatory role of PPARγ was also investigated on
inflammation and oxidative stress in M-HSA-stimulated HUVECs. As
expected, M-HSA resulted in excessive production of TNF-α, IL-1β
and IL-6 in HUVECs, while PPARγ overexpression was capable to
suppress the overproduction of these markers (Fig. 2A-C). Furthermore, elevated levels
of ROS and MDA and a reduced level of SOD were observed in HUVECs
following M-HSA stimulation; these effects were partly reversed by
PPARγ overexpression (Fig. 2D-F).
The aforementioned data indicated a protective role of PPARγ
against M-HSA-stimulated inflammation and oxidative stress in
HUVECs.
PPARγ weakens the activation of ERS in
M-HSA-stimulated HUVECs
Subsequent studies investigated the potential
regulatory mechanism by which ERS is induced by a variety of
physiological and pathological factors including oxidative stress.
The protein expression levels of CHOP, GRP78, p-PERK and p-IRE1α
were significantly increased following M-HSA stimulation,
suggesting that M-HSA triggered the activation of ERS in HUVECs
(Fig. 3). However, this activation
was weakened by PPARγ overexpression, as demonstrated by the
restoration of the protein expression changes following PPARγ
overexpression in M-HSA-stimulated HUVECs.
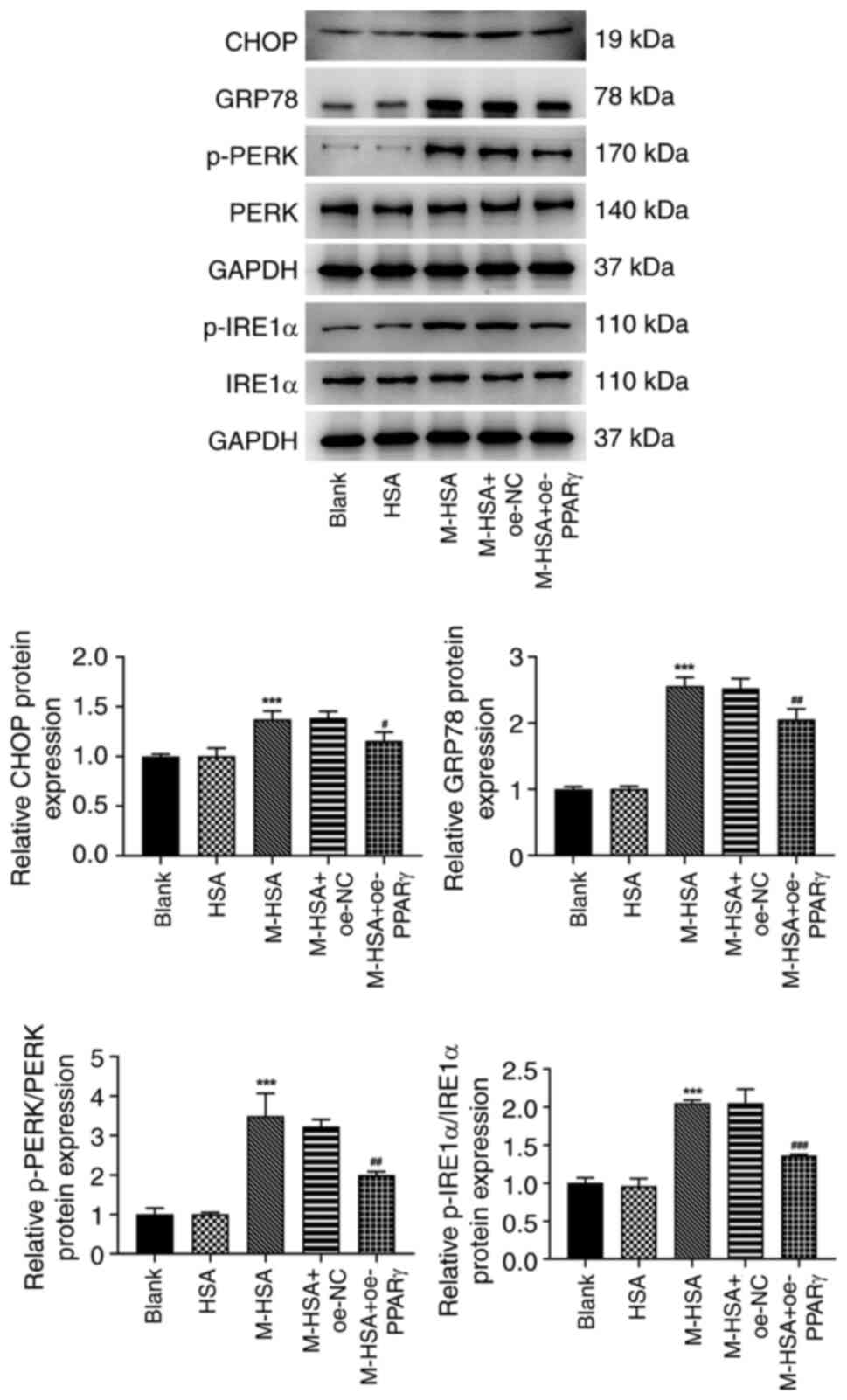 | Figure 3PPARγ weakens the activation of
endoplasmic reticulum stress in M-HSA-stimulated HUVECs. The
protein expression levels of CHOP, GRP78, p-PERK, PERK, IRE1α and
p-IRE1α were detected using western blot analysis.
***P<0.001 vs. HSA; #P<0.05,
##P<0.01 and ###P<0.001 vs. M-HSA +
oe-NC. PPARγ, proliferator-activated receptor γ; M-HSA, modified
glycated human serum albumin; HUVECs, human umbilical vein
endothelial cells; CHOP, C/EBP homologous protein; GRP78,
glucose-regulated protein 78; p, phosphorylated; PERK, protein
kinase (PKR)-like ER kinase; IRE1α, p-inositol requiring enzyme 1α;
oe, overexpressing; NC, negative control. |
TM partly diminishes the effects of
PPARγ on M-HSA-induced endothelial injury in HUVECs
To verify the critical role of ERS during the
regulation of PPARγ in M-HSA-induced HUVECs, the agonist of ERS,
TM, was used and the aforementioned cellular experiments were
re-conducted. It was observed that the inhibitory effects of PPARγ
on M-HSA-induced cell viability loss and cell apoptosis in HUVECs
were weakened by TM (Fig. 4A-C).
Moreover, additional treatment of TM caused a decrease in 6-K-PGF1α
levels and an increase in ET-1 levels compared with the
corresponding levels noted in the M-HSA + oe-PPARγ group (Fig. 4D and E). In addition, the protective effects of
PPARγ against M-HSA-induced inflammation and oxidative stress were
also weakened by TM in HUVECs (Fig.
5A-F). Therefore, these data suggested that the protective role
of PPARγ against M-HSA-induced HUVEC injury could be diminished by
TM, implying that PPARγ may exert its functions by inhibiting the
activation of ERS.
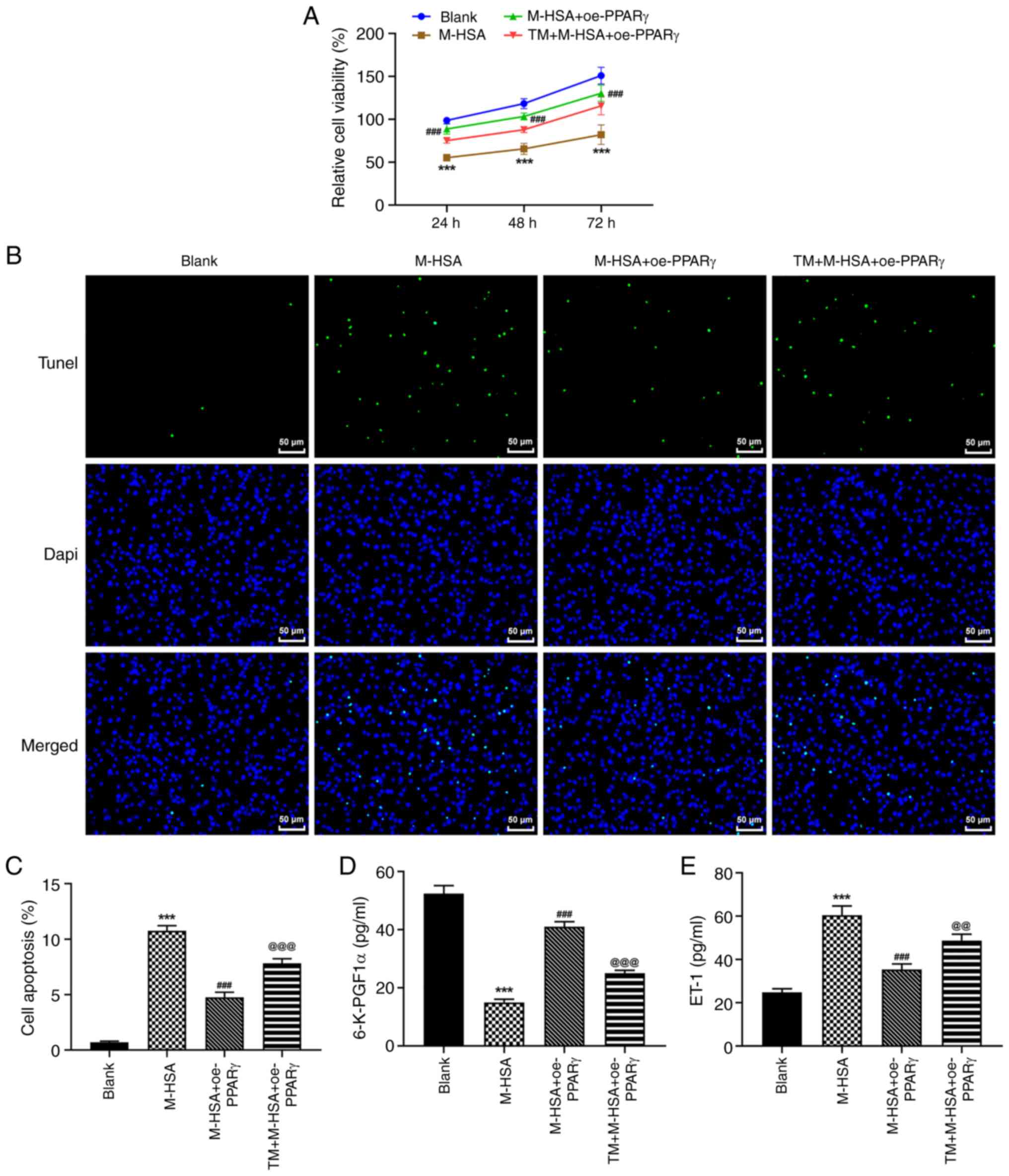 | Figure 4TM partly diminishes the effects of
PPARγ on M-HSA-induced endothelial injury in HUVECs. HUVECs were
stimulated by M-HSA for 24 h to mimic advanced glycation end
products-induced vein thrombosis. Meanwhile, HUVECs were
transfected with oe-PPARγ for 48 h, with or without additional
treatment of TM, an agonist of endoplasmic reticulum stress. (A)
Cell viability was evaluated using Cell Counting Kit-8 assay at
indicated time points (24, 48 and 72 h). (B and C) Cell apoptosis
was determined using TUNEL assay. The concentrations of (D)
6-K-PGF1α and (E) ET-1 were measured by ELISA.
***P<0.001 vs. Blank; ###P<0.001 vs.
M-HSA; @@P<0.01 and @@@P<0.001 vs.
M-HSA + oe-PPARγ. TM, tunicamycin; PPARγ, proliferator-activated
receptor γ; M-HSA, modified glycated human serum albumin; HUVECs,
human umbilical vein endothelial cells; oe, overexpressing;
6-K-PGF1a, 6-keto prostaglandin-F1α; ET-1, endothenin-1. |
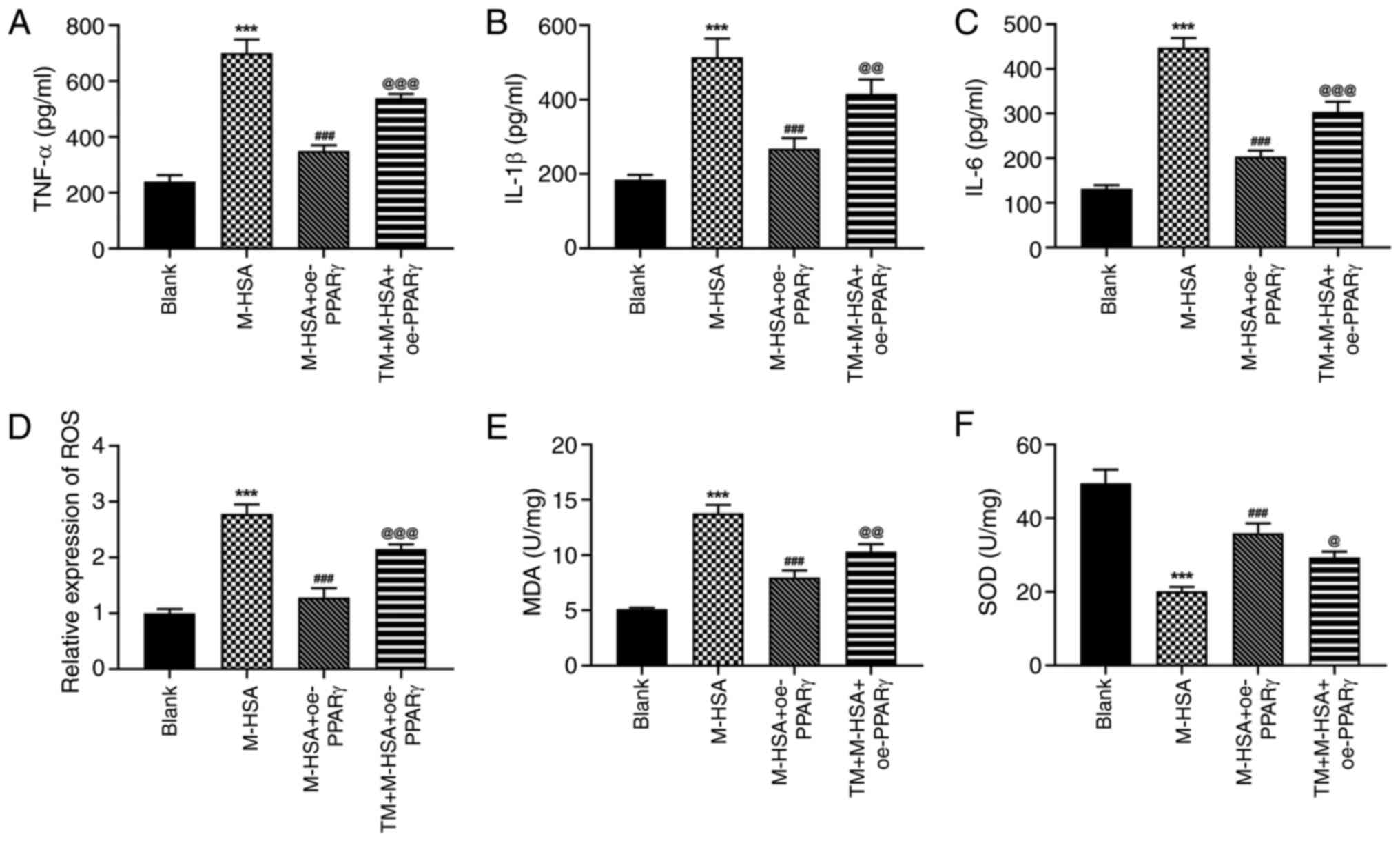 | Figure 5TM partly diminishes the effects of
PPARγ on M-HSA-induced inflammation and oxidative stress in HUVECs.
The production of (A) TNF-α, (B) IL-1β and (C) IL-6 in HUVECs was
measured by ELISA. (D) The level of ROS was detected using CFH-DA
probe. The levels of (E) MDA and (F) SOD in HUVECs were measured
using their corresponding commercial kits. ***P<0.001
vs. Blank; ###P<0.001 vs. M-HSA;
@P<0.05, @@P<0.01 and
@@@P<0.001 vs. M-HSA + oe-PPARγ. TM, tunicamycin;
PPARγ, proliferator-activated receptor γ; M-HSA, modified glycated
human serum albumin; HUVECs, human umbilical vein endothelial
cells; ROS, reactive oxygen species; MDA, malondialdehyde; SOD,
superoxide dismutase; oe, overexpressing. |
Discussion
DVT is recognized as a multifactorial disease
originating from complicated interactions between environmental and
genetic predisposing factors (29). In the present study, the role of
PPARγ in DVT was identified and the regulatory functions and
molecular mechanism of PPARγ were elucidated with regard to
HUVEC-mediated injury. The present study used M-HSA to stimulate
HUVECs so as to mimic AGE-induced DVT. It was determined that PPARγ
was significantly decreased following the stimulation of M-HSA in
HUVECs. Simultaneously, the protective role of PPARγ in
AGEs-induced DVT was verified by its inhibitory effects on cell
apoptosis, endothelial injury, inflammation and oxidative stress in
M-HSA-stimulated HUVECs, illustrating a potential therapeutic
approach against DVT.
The functional capability of the vessel wall
endothelium is essential to maintain vascular function and a
non-thrombotic state. Endothelial dysfunction, which occurs due to
the imbalance between proinflammatory and anti-inflammatory
mediators, oxidative and antioxidant factors, procoagulant and
anticoagulant substances and relaxing and contracting factors,
plays a prominent role in the development of DVT by arousing the
prothrombotic response (10,30-32).
PPARγ is widely expressed in muscle, liver, heart and adipose
tissue, as well as in vascular endothelial and smooth muscle cells
(33). Evidence has shown that
Panax notoginseng saponins-induced activation of PPAR-γ
inhibits thrombin-induced platelet aggregation in vitro and
effectively improves hypercoagulability in vivo (18). The PPARγ agonist rosiglitazone
effectively inhibited inflammation and oxidative stress in injured
HUVECs (34). Notoginsenoside Fc,
a novel triterpenoid derived from P. notoginseng, can
prevent endothelial cell injury via the PPARγ pathway (35). As expected, the present study
demonstrated that PPARγ participated into AGEs-RAGE-triggered
oxidative stress and inflammation during DVT and served as a
protective mediator against the formation of endothelial cell
injury by inhibiting inflammation and oxidative stress.
ERS, also known as the unfolded protein response,
plays an important role in preventing cells against toxic stimuli
or cellular stress-caused deposition of misfolded proteins
(36). Under ERS conditions, GRP78
chaperone binds to misfolded proteins to trigger an adaptive
mechanism via the activation of subsequent signaling pathways,
including PERK, activating transcription factor (ATF) 6α and IRE1α.
Once the unfolded or misfolded proteins are excessive, activated
PERK will phosphorylate eukaryotic initiation factor 2 and further
activate ATF4, which promotes the expression of CHOP and triggers
cell apoptosis (37,38). It has been revealed that AGEs
directly induce ERS in human aortic endothelial cells, playing an
important role in endothelial cell apoptosis (39). As AGE-triggered HUVEC injury
simulates the cellular environment of DVT, it is suggested that ERS
may be involved in the development of DVT. In the present study, an
activation of ERS was found following M-HSA stimulation, as
determined by the upregulation of the protein expression levels of
CHOP, GRP78, p-PERK and p-IRE1α. Simultaneously, PPARγ greatly
suppressed the activation of ERS, which was consistent with
previous studies exploring the regulation of PPARγ on ERS (25,40).
Nevertheless, whether ERS is the cause or the effect of the
regulation of PPARγ during the development of DVT remains unknown;
therefore, the present study addressed this question. Surprisingly,
when TM was employed to promote ERS, the protective function of
PPARγ against inflammation, oxidative stress and apoptosis in
M-HSA-stimulated HUVECs was weakened, demonstrating that ERS is
essential for contributing to HUVEC injury. In addition, PPARγ may
exert its protective role by inhibiting ERS.
However, the present study contains certain
limitations. First, only the regulatory role of PPARγ in
M-HSA-stimulated HUVECs was investigated, which was an in
vitro cell model of DVT. In vivo or clinical studies are
required to verify the findings of the present study. In addition,
more in-depth and comprehensive research is required to elucidate
the molecular mechanism of DVT, so as to discover novel therapeutic
strategies for the clinical treatment of this disease.
In summary, the present study demonstrated that
PPARγ antagonized M-HSA-induced HUVEC injuries by inhibiting cell
apoptosis and balancing thrombosis-related factors, inflammatory
cytokines and oxidative stress-related factors via suppressing the
activation of ERS. Therefore, these findings highlight the
protective role of PPARγ during the development of DVT by
alleviating endothelial injury and imply a promising strategy for
the treatment of DVT.
Supplementary Material
Confirmation of cell transfection
efficacy. Human umbilical vein endothelial cells were transfected
with oe-NC or oe-PPARγ and the protein expression of PPARγ was
examined using western blot analysis. * * *P<0.001
vs. oe-NC. oe, overexpressing; NC, negative control; PPARγ,
proliferatoractivated receptor γ.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by Beijing Jishuitan
Hospital Nova Program (grant no. XKXX202110) and Guangdong Basic
and Applied Basic Research Foundation (grant no.
2019A1515011262).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JLiu conceived and designed the study. YZ, YG, LT,
ML, WJ, XT, PJ, ZC and JLi performed the experiments and collected
the data. YZ, YG, LT and ML analyzed and interpreted the data. YZ
and JLiu wrote and revised the manuscript. YZ and JLiu confirm the
authenticity of all the raw data. All authors read and approved the
final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Konstantinides SV, Torbicki A, Agnelli G,
Danchin N, Fitzmaurice D, Galie N, Gibbs JS, Huisman MV, Humbert M,
Kucher N, et al: 2014 ESC guidelines on the diagnosis and
management of acute pulmonary embolism. Eur Heart J. 35:3033–3069,
3069a-3069k. 2014.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Benjamin EJ, Blaha MJ, Chiuve SE, Cushman
M, Das SR, Deo R, de Ferranti SD, Floyd J, Fornage M, Gillespie C,
et al: Heart disease and stroke statistics-2017 update: A report
from the American heart association. Circulation. 135:e146–e603.
2017.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Huisman MV and Klok FA: Diagnostic
management of acute deep vein thrombosis and pulmonary embolism. J
Thromb Haemost. 11:412–422. 2013.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Fuchs TA, Brill A and Wagner DD:
Neutrophil extracellular trap (NET) impact on deep vein thrombosis.
Arterioscler Thromb Vasc Biol. 32:1777–1783. 2012.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Sun LL, Xiao L, Du XL, Hong L, Li CL, Jiao
J, Li WD and Li XQ: MiR-205 promotes endothelial progenitor cell
angiogenesis and deep vein thrombosis recanalization and resolution
by targeting PTEN to regulate Akt/autophagy pathway and MMP2
expression. J Cell Mol Med. 23:8493–8504. 2019.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Sartori M, Cosmi B, Legnani C, Favaretto
E, Valdré L, Guazzaloca G, Rodorigo G, Cini M and Palareti G: The
wells rule and D-dimer for the diagnosis of isolated distal deep
vein thrombosis. J Thromb Haemost. 10:2264–2269. 2012.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Giordano NJ, Jansson PS, Young MN, Hagan
KA and Kabrhel C: Epidemiology, pathophysiology, stratification,
and natural history of pulmonary embolism. Tech Vasc Interv Radiol.
20:135–140. 2017.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Appelen D, van Loo E, Prins MH, Neumann MH
and Kolbach DN: Compression therapy for prevention of
post-thrombotic syndrome. Cochrane Database Syst Rev.
9(CD004174)2017.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Jin J, Wang C, Ouyang Y and Zhang D:
Elevated miR-195-5p expression in deep vein thrombosis and
mechanism of action in the regulation of vascular endothelial cell
physiology. Exp Ther Med. 18:4617–4624. 2019.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Yang S, Zheng Y and Hou X: Lipoxin A4
restores oxidative stress-induced vascular endothelial cell injury
and thrombosis-related factor expression by its receptor-mediated
activation of Nrf2-HO-1 axis. Cell Signal. 60:146–153.
2019.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Borgel D, Bianchini E, Lasne D, Pascreau T
and Saller F: Inflammation in deep vein thrombosis: A therapeutic
target? Hematology. 24:742–750. 2019.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Li P, Chen D, Cui Y, Zhang W, Weng J, Yu
L, Chen L, Chen Z, Su H, Yu S, et al: Src plays an important role
in AGE-induced endothelial cell proliferation, migration, and
tubulogenesis. Front Physiol. 9(765)2018.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Sena CM, Matafome P, Crisóstomo J,
Rodrigues L, Fernandes R, Pereira P and Seiça RM: Methylglyoxal
promotes oxidative stress and endothelial dysfunction. Pharmacol
Res. 65:497–506. 2012.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Jandeleit-Dahm K and Cooper ME: The role
of AGEs in cardiovascular disease. Curr Pharm Des. 14:979–986.
2008.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Matsui T, Oda E, Higashimoto Y and
Yamagishi S: Glyceraldehyde-derived pyridinium (GLAP) evokes
oxidative stress and inflammatory and thrombogenic reactions in
endothelial cells via the interaction with RAGE. Cardiovasc
Diabetol. 14(1)2015.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Zhang Y, Liu J, Jia W, Tian X, Jiang P,
Cheng Z and Li J: AGEs/RAGE blockade downregulates endothenin-1
(ET-1), mitigating human umbilical vein endothelial cells (HUVEC)
injury in deep vein thrombosis (DVT). Bioengineered. 12:1360–1368.
2021.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Tontonoz P and Spiegelman BM: Fat and
beyond: The diverse biology of PPARgamma. Annu Rev Biochem.
77:289–312. 2008.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Shen Q, Li J, Zhang C, Wang P, Mohammed A,
Ni S and Tang Z: Panax notoginseng saponins reduce high-risk
factors for thrombosis through peroxisome proliferator-activated
receptor-γ pathway. Biomed Pharmacother. 96:1163–1169.
2017.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Yamagishi S, Nakamura K and Matsui T:
Regulation of advanced glycation end product (AGE)-receptor (RAGE)
system by PPAR-gamma agonists and its implication in cardiovascular
disease. Pharmacol Res. 60:174–178. 2009.PubMed/NCBI View Article : Google Scholar
|
|
20
|
El-Bassossy HM, Abo-Warda SM and Fahmy A:
Chrysin and luteolin attenuate diabetes-induced impairment in
endothelial-dependent relaxation: Effect on lipid profile, AGEs and
NO generation. Phytother Res. 27:1678–1684. 2013.PubMed/NCBI View
Article : Google Scholar
|
|
21
|
Xie T, Xu Y, Ji L, Sui X, Zhang A, Zhang Y
and Chen J: Heme oxygenase 1/peroxisome proliferator-activated
receptor gamma pathway protects intimal hyperplasia and mitigates
arteriovenous fistula dysfunction by regulating oxidative stress
and inflammatory response. Cardiovasc Ther.
2022(7576388)2022.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Shou X, Zhou R, Zhu L, Ren A, Wang L, Wang
Y, Zhou J, Liu X and Wang B: Emodin, A Chinese herbal medicine,
inhibits reoxygenation-induced injury in cultured human aortic
endothelial cells by regulating the peroxisome
proliferator-activated receptor-γ (PPAR-γ) and endothelial nitric
oxide synthase (eNOS) signaling pathway. Med Sci Monit. 24:643–651.
2018.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Jin H, Gebska MA, Blokhin IO, Wilson KM,
Ketsawatsomkron P, Chauhan AK, Keen HL, Sigmund CD and Lentz SR:
Endothelial PPAR-γ protects against vascular thrombosis by
downregulating P-selectin expression. Arterioscler Thromb Vasc
Biol. 35:838–844. 2015.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Pathomthongtaweechai N and Chutipongtanate
S: AGE/RAGE signaling-mediated endoplasmic reticulum stress and
future prospects in non-coding RNA therapeutics for diabetic
nephropathy. Biomed Pharmacother. 131(110655)2020.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Yang XL, Mi JH and Dong Q: FABP4
alleviates endoplasmic reticulum stress-mediated
ischemia-reperfusion injury in PC12 cells via regulation of
PPARgamma. Exp Ther Med. 21(181)2021.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Banarjee R, Sharma A, Bai S, Deshmukh A
and Kulkarni M: Proteomic study of endothelial dysfunction induced
by AGEs and its possible role in diabetic cardiovascular
complications. J Proteomics. 187:69–79. 2018.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Jiang Y, Lin L, Liu N, Wang Q, Yuan J, Li
Y, Chung KK, Guo S, Yu Z and Wang X: FGF21 protects against
aggravated blood-brain barrier disruption after ischemic focal
stroke in diabetic db/db male mice via cerebrovascular PPARgamma
activation. Int J Mol Sci. 21(824)2020.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Shi J, Fu C, Su X, Feng S and Wang S:
Ultrasound-stimulated microbubbles inhibit aggressive phenotypes
and promotes radiosensitivity of esophageal squamous cell
carcinoma. Bioengineered. 12:3000–3013. 2021.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Ekim M, Sekeroglu MR, Balahoroglu R, Ozkol
H and Ekim H: Roles of the oxidative stress and ADMA in the
development of deep venous thrombosis. Biochem Res Int.
2014(703128)2014.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Poredos P and Jezovnik MK: Endothelial
dysfunction and venous thrombosis. Angiology. 69:564–567.
2018.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Kirwan CC, McCollum CN, McDowell G and
Byrne GJ: Investigation of proposed mechanisms of
chemotherapy-induced venous thromboembolism: Endothelial cell
activation and procoagulant release due to apoptosis. Clin Appl
Thromb Hemost. 21:420–427. 2015.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Stein-Merlob AF, Hara T, McCarthy JR,
Mauskapf A, Hamilton JA, Ntziachristos V, Libby P and Jaffer FA:
Atheroma susceptible to thrombosis exhibit impaired endothelial
permeability in vivo as assessed by nanoparticle-based fluorescence
molecular imaging. Circ Cardiovasc Imaging.
10(e005813)2017.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Sigmund CD: Endothelial and vascular
muscle PPARgamma in arterial pressure regulation: Lessons from
genetic interference and deficiency. Hypertension. 55:437–444.
2010.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Xu L, Zhao G, Zhu H, Wang S, Sun A, Zou Y
and Ge J: Peroxisome proliferator-activated receptor-γ antagonizes
LOX-1-mediated endothelial injury by transcriptional activation of
miR-590-5p. PPAR Res. 2019(2715176)2019.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Liu J, Jiang C, Ma X and Wang J:
Notoginsenoside Fc attenuates high glucose-induced vascular
endothelial cell injury via upregulation of PPAR-γ in diabetic
sprague-dawley rats. Vascul Pharmacol. 109:27–35. 2018.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Wu J and Kaufman RJ: From acute ER stress
to physiological roles of the unfolded protein response. Cell Death
Differ. 13:374–384. 2006.PubMed/NCBI View Article : Google Scholar
|
|
37
|
He Z, Wang M, Zhao Q, Li X, Liu P, Ren B,
Wu C, Du X, Li N and Liu Q: Bis(ethylmaltolato)oxidovanadium (IV)
mitigates neuronal apoptosis resulted from amyloid-beta induced
endoplasmic reticulum stress through activating peroxisome
proliferator-activated receptor gamma. J Inorg Biochem.
208(111073)2020.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Hammadi M, Oulidi A, Gackiere F,
Katsogiannou M, Slomianny C, Roudbaraki M, Dewailly E, Delcourt P,
Lepage G, Lotteau S, et al: Modulation of ER stress and apoptosis
by endoplasmic reticulum calcium leak via translocon during
unfolded protein response: Involvement of GRP78. FASEB J.
27:1600–1609. 2013.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Adamopoulos C, Farmaki E, Spilioti E,
Kiaris H, Piperi C and Papavassiliou AG: Advanced glycation
end-products induce endoplasmic reticulum stress in human aortic
endothelial cells. Clin Chem Lab Med. 52:151–160. 2014.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Chi X, Jiang Y, Chen Y, Yang F, Cai Q, Pan
F, Lv L and Zhang X: Suppression of microRNA27a protects against
liver ischemia/reperfusion injury by targeting PPARgamma and
inhibiting endoplasmic reticulum stress. Mol Med Rep. 20:4003–4012.
2019.PubMed/NCBI View Article : Google Scholar
|