Introduction
Continuous improvements in living standards and
increasing average lifespan have led to a corresponding rise in the
incidence of neurodegenerative diseases, for which aging is the
primary risk factor (1).
Alzheimer's disease (AD), the most common type of dementia, is a
progressive neurodegenerative disease among the elderly population
(2). It is estimated that by
mid-century, the number of Americans aged ≥65 years with AD may
grow to 13.8 million (3). This
represents a steep increase from the estimated 5.8 million
Americans aged ≥65 years who have AD today (3). Patients diagnosed with AD experience
gradual deterioration of memory and cognitive function, accompanied
by a range of behavioral disturbances and neuropsychiatric symptoms
(4,5).
Pathological characteristics of AD involve numerous
aspects, such as tau phosphorylation, and abnormal expression of
glycogen synthase kinase 3β (GSK3β), synaptophysin, brain-derived
neurotrophic factor (BDNF) and inflammatory cytokines (6-10).
Another notable characteristic is excessive amyloid β (Aβ)
accumulation outside neurons, eventually resulting in the formation
of plaques (11). This phenomenon
has long been understood to indicate that abnormal amyloidogenic
processing is a significant element of AD development (12). However, research has suggested that
numerous elderly individuals have Aβ deposits in their brains
without exhibiting AD symptoms, suggesting that the deposition of
Aβ plaques is not a sufficient condition for the occurrence of AD
(13). By contrast, the
distribution and severity of neurofibrillary tangles appear to have
a stronger association with cognitive dysfunction than Aβ plaque
deposition (14). At present, no
successful therapy is available that can cure or significantly slow
AD progression, despite extensive research on the etiology of the
disorder and considerable investment from the pharmaceutical
industry (15). As a result, the
underlying mechanisms of AD must be elucidated more comprehensively
to develop viable treatment strategies.
Numerous bioactive compounds derived from natural
sources have exhibited potential anti-AD effects in both laboratory
and clinical studies, such as tanshinone IIA (16), naringin (17) and ellagic acid (18). Magnolia officinalis
(commonly termed Houpu Magnolia or Magnolia-bark), a species of the
genus Magnolia belonging to the family Magnoliaceae, is an
endangered deciduous tree in China (19). Magnolol, a bioactive component
isolated from Magnolia officinalis, has been extensively
documented to possess diverse biological properties, including
anti-oxidative, anticancer and anti-inflammatory effects. For
example, magnolol has been shown to possess potential in mitigating
oxidative stress in white adipocytes, and is thus being considered
a promising strategy for combating obesity (20). It has the ability to inhibit the
proliferative and migratory capacities of diverse types of human
cancer, including pancreatic cancer (21), cervical cancer (22) and hepatocellular carcinoma
(23). Furthermore, the
administration of magnolol has been shown to confer advantages in
attenuating inflammation in patients with diabetic periodontitis
(24). Recently, the
neuroprotective potential of magnolol in AD-related pathology has
also been uncovered. Wang and Jia (25) reported that magnolol can improve
cognitive decline through regulating autophagy and the
AMPK/mTOR/ULK1 pathway. Chen et al (26) established a mouse model of AD with
brain insulin resistance, and demonstrated that magnolol could
interact with microRNA-200c to alleviate neuroinflammation. Zhu
et al (27) found that the
magnolol-mediated cAMP/PKA/CREB pathway could significantly
mitigate Aβ-induced neuronal injury in SH-SY5Y neuroblastoma cells.
It is widely recognized that the initiation and progression of AD
involve a multifaceted biological process encompassing various
regulatory mechanisms (28,29).
The potential existence of additional regulatory mechanisms
underlying the impact of magnolol on the progression of AD is a
topic of great interest to researchers.
In the present study, a preliminary investigation
was conducted into the effects of varying doses of magnolol on
several factors related to AD pathology, including tau
phosphorylation, GSK3β, synaptophysin, BDNF, inflammatory
cytokines, as well as the activation of astrocytes and microglia.
The present study aims to further enhance the understanding of AD
pathogenesis and establish a robust foundation for targeted AD
therapy.
Materials and methods
Reagents and antibodies
Magnolol (analytical standard, purity ≥98%) was
provided by Shanghai Yuanye Biotechnology Co., Ltd. Congo red dye
and Aβ1-42 were acquired from MilliporeSigma, while the
hematoxylin dye was supplied by Beyotime Institute of
Biotechnology. Goat serum was obtained from Gibco; Thermo Fisher
Scientific, Inc. RIPA lysis buffer, bovine serum albumin, ECL Kit
and BCA Protein Assay Kit were obtained from Thermo Fisher
Scientific, Inc. The QIAwave RNA Mini Kit, First Strand Kit and
QuantiFast SYBR® Green PCR Kit were obtained from Qiagen
GmbH. The primary antibodies used in the present study were IL-6
(Abcam; cat. no. ab290735), IL-1β (Abcam; cat. no. ab315084), tau
(Abcam; cat. no. ab254256), phosphorylated (p-)tau (Invitrogen;
Thermo Fisher Scientific, Inc.; cat. no. MN1020), GSK3β (Abcam;
cat. no. ab32391), p-GSK3β (Abcam; cat. no. ab75814), synaptophysin
(Proteintech Group, Inc.; cat. no. 17785-1-AP), BDNF (Proteintech;
cat. no. 28205-1-AP), glial fibrillary acidic protein (GFAP;
Proteintech Group, Inc.; cat. no. 16825-1-AP), ionized calcium
binding adaptor molecule 1 (Iba1; Proteintech Group, Inc.; cat. no.
10904-1-AP) and GAPDH (Proteintech Group, Inc.; cat. no.
60004-1-Ig). The HRP-conjugated anti-rabbit (cat. no. A21020) and
HRP-conjugated anti-mouse (cat. no. A21010) secondary antibodies
were obtained from Abbkine Scientific Co., Ltd. FITC-labeled goat
anti-mouse secondary antibody (cat. no. ab6785) was obtained from
Abcam.
Animal grouping and treatment
Healthy male C57BL/6 mice (age, 10-12 weeks; weight,
25-30 g) with normal locomotor and cognitive functions were
purchased from Charles River Laboratories, Inc. Before grouping the
mice, visible-platform training was carried out to determine the
baseline differences in vision and motivation. Mice with abnormal
locomotor abilities were excluded. Subsequently, mice were housed
in specific pathogen-free cages under standard laboratory
conditions including 22-25˚C temperature, 40-55% relative humidity
and 12-h light/dark cycle, with free access to water and food. The
Aβ1-42 stock solution was prepared at a concentration of
1 mg/ml in sterile PBS and then incubated at 37˚C for 5 days to
facilitate oligomerization.
A total of 30 C57BL/6 mice were randomly divided
into five groups (n=6 mice/group): i) Control; ii) AD model; iii) 5
mg/kg magnolol + AD model; iv) 10 mg/kg magnolol + AD model; and v)
20 mg/kg magnolol + AD model. All surgical procedures were
performed under aseptic conditions. The AD model was established as
follows: The mice were anesthetized by intraperitoneal (i.p.)
injection of pentobarbital sodium (50 mg/kg), and then subsequently
secured onto a brain stereotaxic device in a prone position. As
previously described (30,31), the hippocampal CA1 region serves a
critical role in cognitive functions such as learning and memory,
and damage or lesions in this region are closely related to
cognitive impairment in AD. Additionally, bilateral injection can
simulate the pathological changes of diseases more comprehensively,
improving the stability and reliability of the model (32). Therefore, the bilateral hippocampal
CA1 region was selected as the targeted injection area. The
specific coordinate position was 2.4 mm dorsoventral, 0.2 mm
anteroposterior and 1 mm mediolateral to the bregma. The aggregated
form of Aβ1-42 was administered via
intracerebroventricular injection at a volume of 3 µl and a speed
of 1 µl/min. The injector was left in place for 5 min. Mice in the
control group received an equivalent amount of normal saline. To
consider animal welfare, animal distress was alleviated as much as
possible. A 0.2-ml dose of gentamicin was injected to the suture
site for anti-inflammatory treatment when the incision was sutured.
Meanwhile, the animals were housed individually and permitted to
recuperate from the anesthesia on a heated mat in order to regulate
body temperature at 37.5±0.5˚C. After 7 days, the treatment groups
were administered varying doses of magnolol (5, 10 and 20 mg/kg) by
gavage every day, while mice in the control and AD model groups
received an equal volume of normal saline. During the experiments,
changes in body weight were monitored once a week, and changes in
food and water intake were observed. Although it was not expected,
a rapid decrease in normal body weight >20% was defined as a
humane endpoint for the present study. In this study, none of the
mice reached the humane endpoint.
A Morris water maze (MWM) test was conducted after 2
months of magnolol treatment. No animals died naturally during the
experiments, and all mice were euthanized by overdose with
pentobarbital sodium (200 mg/kg; i.p.). The death was confirmed by
cardiac and respiratory arrest, along with the absence of response
to tail clamping. A total of 3 mice were randomly selected from
each group, and the hippocampal tissues were collected for Congo
red and immunofluorescence staining. The hippocampal tissues of the
remaining mice were stored at -80˚C until use. The whole experiment
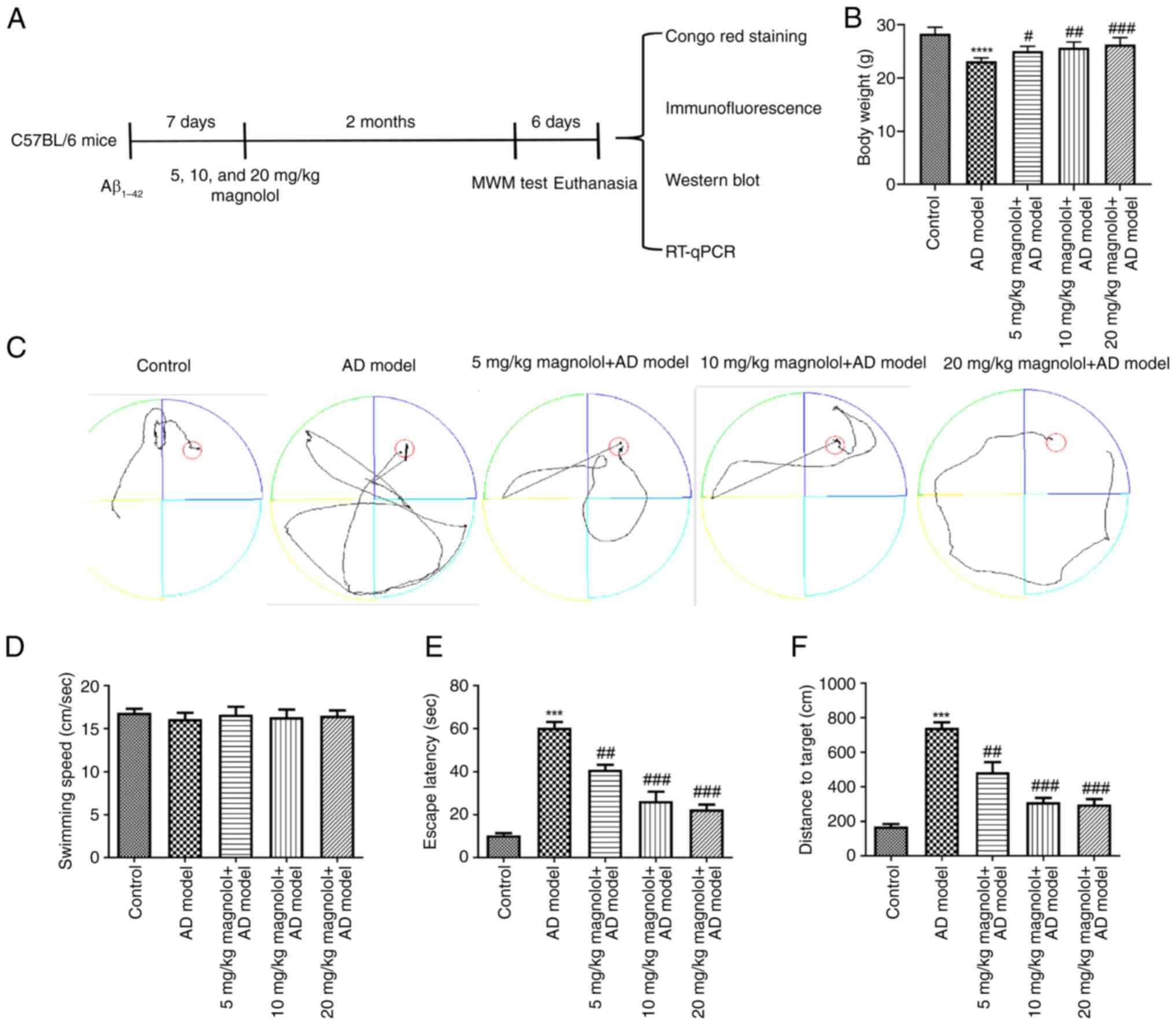
lasted 73 days and a schematic diagram of the experimental protocol
is shown in Fig. 1A. The animal
experiments were conducted in compliance with the guidelines
outlined in the National Institutes of Health Guide for the Care
and Use of Laboratory Animals (33) and received approval from The Ethics
Committee of The School of Medicine of Jinhua Polytechnic (Jinhua,
China; approval no. AL-JSYJ202334).
MWM test
As previously described (25), the MWM test was applied to evaluate
the spatial memory of the mice. A circular pool with a height of 50
cm and a diameter of 150 cm was used. The water was filled to a
depth of 30 cm, while maintaining a temperature of 24˚C. The
swimming pool was artificially partitioned into four quadrants,
designated as quadrant I, II, III and IV. During the 5-day spatial
navigation test, a round platform with a diameter of 10 cm was
positioned at the center of quadrant IV and submerged 1 cm below
the water surface, followed by removing it when the 1-day probe
task was conducted. The mice were each subjected to two tests per
day, with a minimum interval of 15 min between tests. Each mouse
was introduced into the pool and given 60 sec to seek the platform.
The time taken to locate the concealed platform was recorded as
escape latency (sec). On day 6, the hidden platform was removed to
evaluate the memory ability. Subsequently, the mice were permitted
to swim freely for 60 sec to seek the removed platform. A video
tracking system (version 3.0; SMART, Panlab) was employed to record
the distance to target, escape latency and swimming speed.
Section preparation
The hippocampal tissues were collected and promptly
fixed in 4% paraformaldehyde (PFA) at 4˚C overnight. Subsequently,
the specimens were immersed in a solution of 30% sucrose-PFA until
they sank to the bottom. The samples were then embedded in paraffin
and flash-frozen using liquid nitrogen. Slices with a thickness of
4 µm were obtained using a cryostat (Leica Microsystems GmbH),
stored at -80˚C, and used for Congo red and immunofluorescence
staining.
Congo red staining
Congo red staining was used for Aβ plaque
visualization in the CA1 region of the hippocampus, as previously
reported (34,35). The sections underwent
deparaffinization and hydration with distilled water, followed by
staining in a solution of 0.5% Congo red for 20 min at room
temperature. Subsequently, the sections were rinsed in distilled
water and rapidly differentiated in an alkaline alcohol solution.
The sections were then washed in tap water for 1 min, followed by
counterstaining with hematoxylin for 30 sec at room temperature and
subsequent rinsing again in tap water for 2 min. Finally, the
sections were dehydrated and mounted using resinous mounting
medium. For semi-quantification of Congo red staining, images were
captured from three hippocampal sections per sample, and Congo
red-positive cells were counted manually from the CA1 region of the
hippocampus using five random regions, under an optical microscope
(scale bar, 50 µm; Olympus Corporation). The number of Aβ plaques
was represented by the number of Congo red-positive cells.
Immunofluorescence staining
The tissue slices underwent deparaffinization in
xylene for 10 min at room temperature and rehydration with
descending concentrations of ethanol (100, 95 and 70% for 3-5 min
each), followed by antigen retrieval in heated sodium citrate at
80˚C for 25 min. Subsequently, the slices were blocked with 4% goat
serum for 15 min at room temperature, after which, the slices were
incubated overnight at 4˚C with the following primary antibodies:
IL-1β, IL-6, p-tau, Iba1 and GFAP (all diluted to 1:50). After
three washes with PBS, the slices were incubated with FITC-labeled
goat anti-mouse secondary antibody (1:200) for 1 h in the dark at
room temperature. DAPI was used for nuclear staining at room
temperature for 15 min. For the expression determination, images
were captured from three hippocampal sections per sample. The
IL-1β-, IL-6-, p-tau-, Iba1- and GFAP-positive cells were evaluated
under a fluorescence microscope (Olympus Corporation; scale bar,
100 µm) from five different CA1 regions of hippocampus.
Image-Pro-Plus (version 6.0; Media Cybernetics) was adopted to
analyze the fluorescence intensity.
Western blotting
Proteins were extracted from hippocampal tissues
using RIPA lysis buffer with protease inhibitors, and their
concentration was determined using the BCA Protein Assay Kit. A
total of ~25 µg proteins were separated by SDS-PAGE on a 10% gel
and subsequently transferred to a PVDF membrane. The membrane was
then blocked with 5% bovine serum albumin for 2 h at room
temperature before being incubated overnight at 4˚C with primary
antibodies against tau (1:1,000), p-tau (1:1,000), GSK3β (1:5,000),
p-GSK3β (1:5,000), synaptophysin (1:20,000), BDNF (1:1,000), GFAP
(1:5,000), Iba1 (1:1,000) and GAPDH (1:50,000). After washing the
membranes three times with Tris-buffered saline-Tween-20 (0.05%),
the HRP-conjugated secondary antibodies (1:10,000) were added and
incubated at room temperature for 1 h. The internal reference used
was GAPDH. Finally, the immunoreactive protein bands were
visualized using an ECL Kit under a Gel-Proanalyzer (version 4.0;
MilliporeSigma).
Isolation of RNA and reverse
transcription-quantitative PCR (RT-qPCR)
The QIAwave RNA Mini Kit was employed for RNA
isolation. Subsequently, cDNA was synthesized using the First
Strand Kit according to the manufacturer's protocol, and subjected
to qPCR analysis using a QuantiFast SYBR Green PCR Kit on the
Real-time PCR System (Applied Biosystems; Thermo Fisher Scientific,
Inc.). The thermocycling conditions were as follows: 95˚C for 10
min, followed by 40 cycles at 94˚C for 10 sec, 60˚C for 20 sec and
72˚C for 34 sec. Gene expression levels were determined using the
2-ΔΔCq method (36)
with GAPDH used as the internal control. The gene primers utilized
in this study are detailed in Table
I.
 | Table IReverse transcription-quantitative
PCR primer sequences. |
Table I
Reverse transcription-quantitative
PCR primer sequences.
| Gene | Sequence
(5'-3') |
|---|
| Synaptophysin | Forward:
GACGTTGGTAGTGCCTGTGA |
| | Reverse:
GCACAGGAAAGTAGGGGGTC |
| BDNF | Forward:
CGGAGAGCAGAGTCCATTCAG |
| | Reverse:
CCAGTATACCAACCCGGAGC |
| GFAP | Forward:
CCTGCCAGCTCTCCCT |
| | Reverse:
AAAGGTGTGGCTGAAATGCG |
| Iba1 | Forward:
TGAGGAGATTTCAACAGAAGCTGA |
| | Reverse:
AGACGCTGGTTGTCTTAGGC |
| GAPDH | Forward:
AAGAGGGATGCTGCCCTTAC |
| | Reverse:
TACGGCCAAATCCGTTCACA |
Statistical analysis
The data analysis was conducted using SPSS software
version 20.0 (IBM Corp.) and the results are presented as the mean
± standard deviation. Statistical differences among the groups were
determined using one-way ANOVA, followed by Tukey's multiple
comparisons post hoc test. P<0.05 was considered to indicate a
statistically significant difference.
Results
Treatment with magnolol mitigates
cognitive impairment in Aβ1-42-induced mice
The body weight of mice in different groups was
monitored. As shown in Fig. 1B,
compared with that in the control group, the body weight of mice
with AD was significantly decreased; however, compared with in the
AD model group, administration of different doses of magnolol,
particularly 20 mg/kg, significantly restored the body weight. The
track plots of mice in the MWM test are shown in Fig. 1C. First, the speed of mice swimming
in the water maze was assessed to exclude false-positive results.
The results revealed that there was no significant difference in
the swimming speed of each group of mice (Fig. 1D). Next, it was revealed that the
AD model group spent more time to reach the platform compared with
the control group in the spatial navigation test (Fig. 1E). However, administration of
different doses of magnolol (5, 10 and 20 mg/kg) significantly
ameliorated the effects of Aβ1-42 on escape latency. In
the spatial probe test, the memory retention of the platform
location performed on the day following the spatial navigation test
was assessed. It was shown that compared with the mice in the AD
model group, magnolol-treated AD mice showed improved spatial
learning and memory ability. This was reflected by significant
progressive reductions in distance traveled to the target platform
(Fig. 1F).
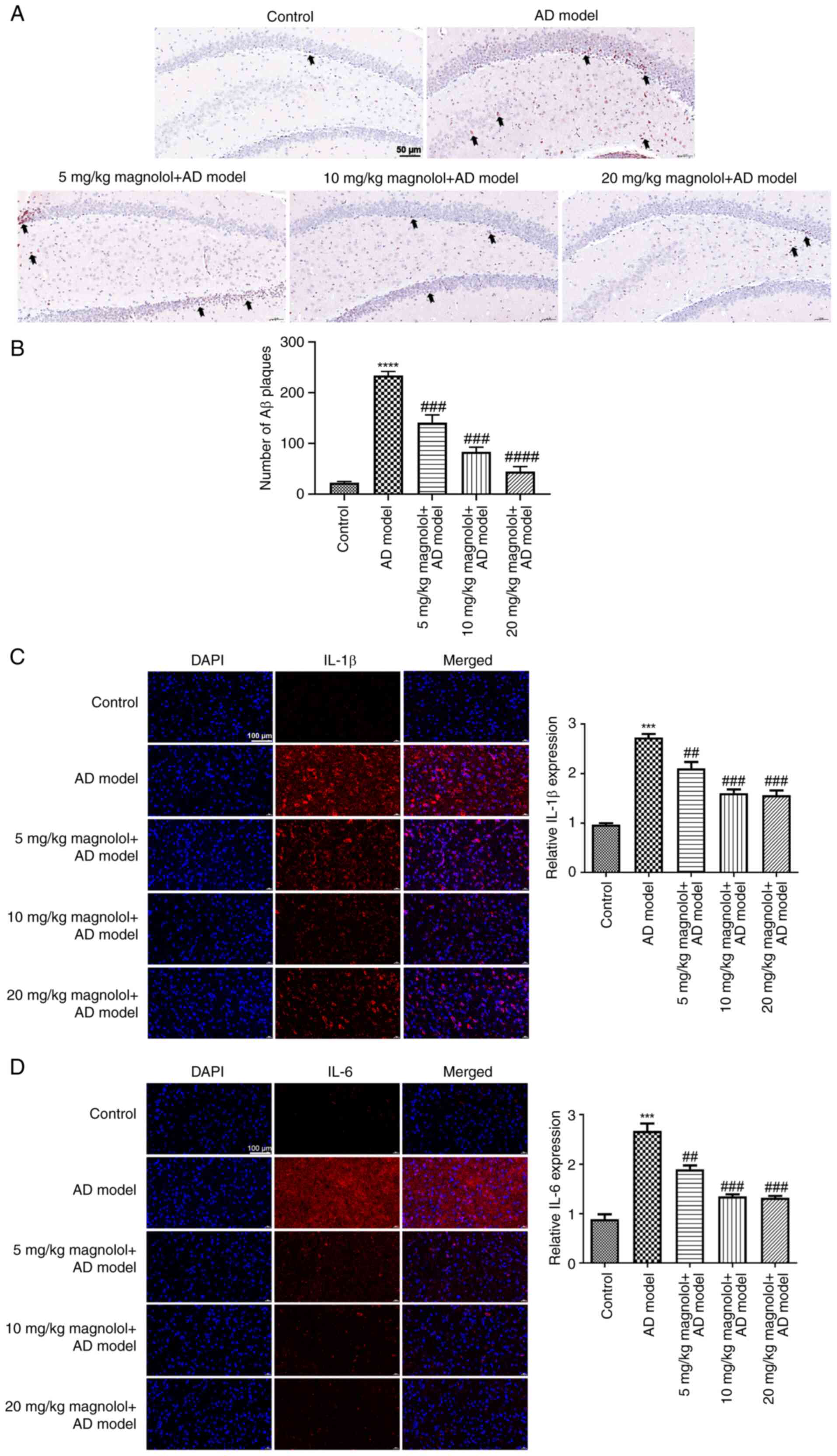
Magnolol reduces Aβ plaque deposits
and inhibits inflammation in Aβ1-42-induced mice
The accumulation of Aβ plaques is a crucial
characteristic feature of AD. Therefore, the hippocampal tissues
were subjected to Congo red staining for Aβ plaque deposition
detection. Sections with Aβ plaque accumulation displayed a
red-colored Congo stain in the CA1 region of the hippocampus. As
shown in Fig. 2A, almost no Congo
red-positive cells were observed in the control group, indicating
that there was almost no accumulation of plaques in the hippocampal
CA1 region of the control mice. Hippocampal sections from
Aβ1-42-induced mice in the model group showed more dense
Congo red stains, indicating that Aβ plaques had accumulated.
Administration of magnolol, specifically at a dosage of 20 mg/kg,
notably reduced the number of Congo red-positive cells in
hippocampal tissues and indicated that treatment with magnolol
significantly inhibited the accumulation of Aβ plaques (Fig. 2B). After activation with plaque
accumulation, astrocytes and microglia release a diverse array of
pro-inflammatory cytokines that lead to impaired neuronal function
in the hippocampus, which is crucial for learning and memory
formation (37).
Immunofluorescence staining was conducted to evaluate the
expression of IL-1β and IL-6 in the hippocampus. It was indicated
that relative expression levels of IL-1β and IL-6 were notably
enhanced in the model group, which was in contrast to the control
group (Fig. 2C and D). However, treatment with varying doses
of magnolol significantly decreased IL-1β and IL-6 levels in the
hippocampus of Aβ1-42-induced mice.
Magnolol decreases the expression of
tau, p-tau, GFAP and Iba1, and restores the levels of GSK3β,
p-GSK3β, synaptophysin and BDNF
The occurrence of AD involves numerous crucial
pathological mechanisms, such as the formation of intracellular
neurofibrillary tangles, disrupted synaptic plasticity, and the
activation of astrocytes and microglia (38-40).
Therefore, an investigation was conducted into the expression of
pertinent factors. As illustrated in Fig. 3A, in comparison with the control
group, the protein levels of tau, p-tau, GFAP and Iba1 were
increased in the model group, whereas GSK3β, p-GSK3β, synaptophysin
and BDNF levels were decreased. However, treatment with magnolol,
specifically at a dosage of 10 or 20 mg/kg, significantly decreased
the protein levels of tau, p-tau, GFAP and Iba1, and restored the
levels of GSK3β, p-GSK3β, synaptophysin and BDNF in
Aβ1-42-induced mice. RT-qPCR analysis detected the mRNA
expression levels of synaptophysin, BDNF, GFAP and Iba1 (Fig. 3B), while immunofluorescence
staining assessed p-tau, Iba1 and GFAP expression (Fig. 3C and D), and the results provided further
validation for the aforementioned findings.
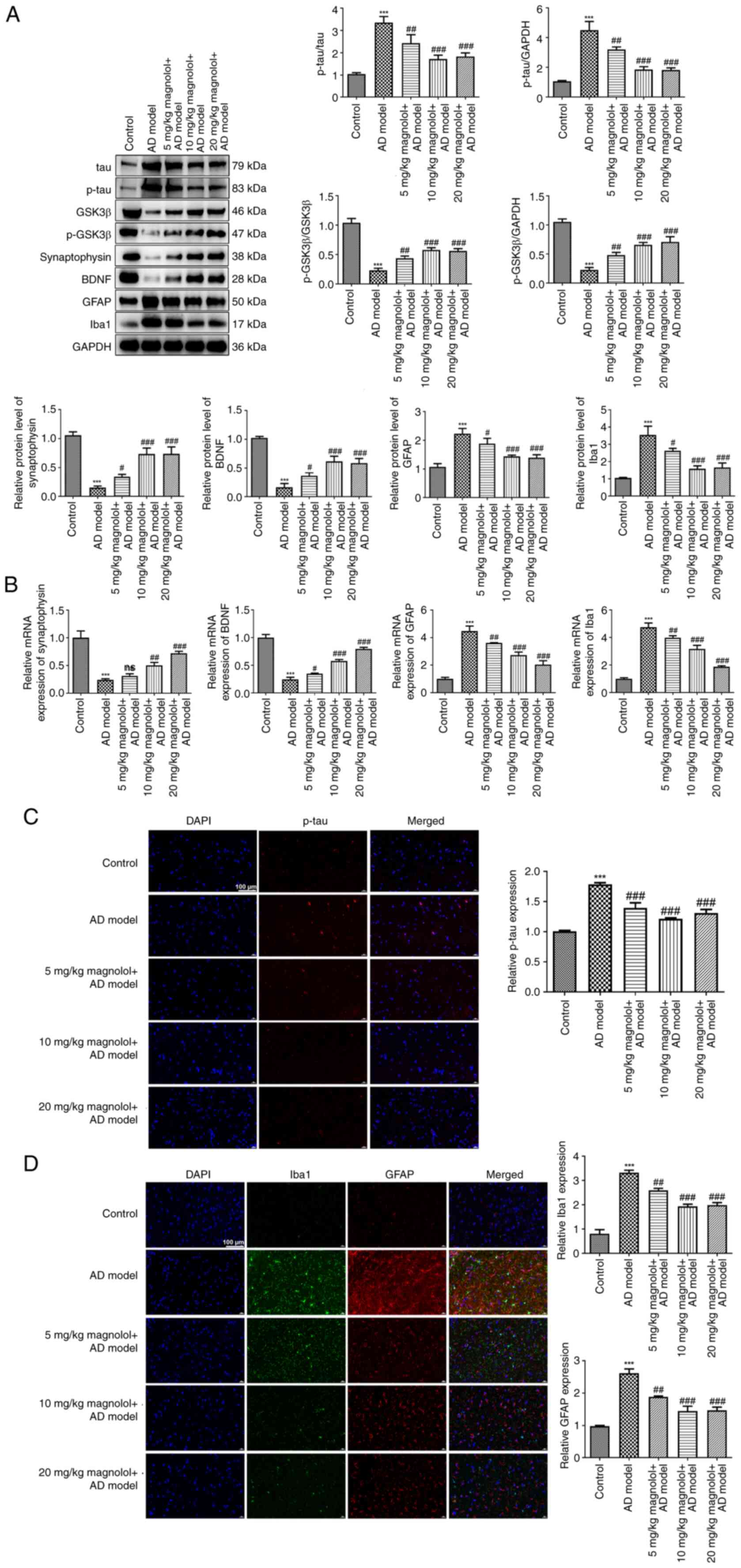 | Figure 3Magnolol decreases the expression of
tau, p-tau GFAP and Iba1, and restores the levels of GSK3β,
p-GSK3β, synaptophysin and BDNF. (A) Protein levels of tau, p-tau,
GSK3β, p-GSK3β, synaptophysin, BDNF, GFAP and Iba1 in each group
were determined by western blotting. (B) mRNA expression levels of
synaptophysin, BDNF, GFAP and Iba1 in each group were detected by
reverse transcription-quantitative PCR. Expression levels of (C)
p-tau, (D) Iba1 and GFAP in each group were analyzed by
immunofluorescence staining (scale bar, 100 µm).
***P<0.001 vs. control; #P<0.05,
##P<0.01, ###P<0.001 vs. AD model. ns,
not significant; AD, Alzheimer's disease; GSK3β, glycogen synthase
kinase 3β; BDNF, brain-derived neurotrophic factor; GFAP, glial
fibrillary acidic protein; Iba1, ionized calcium binding adaptor
molecule 1; p, phosphorylated. |
Discussion
Aβ1-42 has been widely employed to induce
animal models of AD due to its neurotoxic effects, including
disruption of calcium homeostasis, neuroinflammation, induction of
cell death and mitochondrial dysfunction (41). As previously described (42,43),
the dosage of Aβ administered in the present study elicits a
comprehensive spectrum of AD symptoms within 7 days. The hallmark
manifestations of AD encompass cognitive decline and memory loss.
Previous research has frequently utilized the MWM test to evaluate
long-term spatial memory (44,45).
In the present study, the findings indicated that magnolol-treated
mice with AD exhibited notable improvements in cognitive function,
as evidenced by a significant reduction in the distance to the
target and escape latency assessed using the MWM test. These
findings provide evidence that magnolol is an effective bioactive
compound for treating AD. There is mounting evidence to indicate
that the excessive accumulation of Aβ in patients with AD or in the
APP/PS1 mouse model is strongly associated with cognitive
impairment (46,47). The results of the present study
lend credence to the previous study (48) suggesting that
Aβ1-42-induced AD mice showed a significant accumulation
of Aβ plaques. Notably, 20 mg/kg magnolol significantly reduced the
formation of Aβ plaques. Additionally, compelling evidence suggests
that inflammation caused by Aβ may lead to cell apoptosis and
impaired neuronal function, playing a crucial role in the
pathogenesis of AD (49).
Therefore, as a potent therapeutic agent for AD, magnolol may also
exhibit anti-inflammatory properties during AD progression. As
hypothesized, the results of the present study confirmed that the
administration of magnolol effectively inhibited the expression of
inflammatory cytokines (IL-1β and IL-6) in the hippocampus of
Aβ1-42-induced mice. These findings may provide evidence
supporting the previous results that magnolol can effectively
alleviate the symptoms of AD by inhibiting Aβ deposits and
neuroinflammation.
The development of AD also involves a multitude of
other crucial pathological mechanisms, including the formation of
intracellular neurofibrillary tangles, compromised synaptic
plasticity, and the activation of astrocytes and microglia
(38-40).
Tau, accounting for 80% of the proteins associated with
microtubules, serves a crucial role in maintaining the stability of
axonal transport tracks and microtubules, while
hyperphosphorylation of tau represents a primary factor
contributing to the formation of neurofibrillary tangles (50). Tau can undergo phosphorylation at
several AD-related sites through the activation of GSK3β, which is
one of the direct causes of tau hyperphosphorylation and can
eventually cause spatial memory deficit (51). At the neuronal level, it has been
documented that the absence of GSK3β may lead to a decrease in the
stability of dendritic spines and disrupted synaptic transmission
within a particular region of the hippocampus (CA1 subset) and
cortices, potentially resulting in adverse effects during the
progression of AD (52). Learning
and memory in the central nervous system rely on synaptic
plasticity at a molecular level, and abnormal synaptic function is
closely linked to cognitive decline in AD (53). The loss of GSK3β in the dentate
gyrus of mice has been shown to inhibit synaptic transmission in
the hippocampus through reducing synaptophysin expression (54). As a member of the neurotrophin
family, elevated levels of BDNF have been reported to be associated
with synaptic transmission and synaptic plasticity (55). Furthermore, neurodegenerative
conditions, including AD, are associated with the activation of
astrocytes and microglia. However, research has shown that
inhibiting their activities can effectively mitigate
neurodegeneration (56). In the
present study, given the inhibitory role of magnolol against Aβ
deposits and neuroinflammation in Aβ1-42-induced mice,
the effects of magnolol on these pathological processes were
further evaluated by assessing the levels of associated proteins or
markers. It was demonstrated that moderate and high doses of
magnolol exhibited a more potent inhibitory effect on the levels of
tau and p-tau, while also leading to a more pronounced restoration
of GSK3β and p-GSK3β expression. The findings indicated that
magnolol may mitigate the progression of AD by inhibiting the
accumulation and hyperphosphorylation of tau as well as activating
GSK3β. Concurrently, the administration of magnolol was found to
increase synaptophysin and BDNF levels, indicating that magnolol
may exert a reparative effect on synaptic plasticity. Moreover, by
reducing the levels of activated astrocyte-specific marker (GFAP)
and activated microglia-specific marker (Iba1), it was indicated
that magnolol could effectively suppress the activation of
astrocytes and microglia, thereby mitigating the development of
AD.
The present study has certain limitations. First,
experiments on the role of magnolol in in vitro experiments,
such as in immortalized mouse hippocampal cells, are needed.
Second, elaborating on the inter-relationships between
proteins/markers related to key pathological mechanisms of AD and
how magnolol affects the interactions between these mechanisms in
the present study could be more rigorous. To attain this goal, more
elaborate experiments are required. For example, to evaluate how
magnolol affects the activation of microglia and astrocytes,
double-labeling immunofluorescence analyses of Aβ deposits and
microglia marked with IL-1β and CD11b, as well as double-labeling
immunofluorescence analyses of Aβ deposits and astrocytes marked by
GFAP should be performed. In addition, immunofluorescence staining
for microtubule associated protein 2 and Golgi staining for the
detection of dendritic spine densities in the CA1 region could be
used to assess synaptic plasticity. Third, aside from
Aβ1-42-injected AD mouse models, genetic models of AD,
such as APP/PS1 transgenic mice, is another important animal model
to study the pathological mechanism of AD. Future studies aim to
investigate the more precise and detailed mechanisms of magnolol in
this transgenic mouse model. Furthermore, there were some
limitations with the current experimental method. First,
introducing well-known positive drugs for treating memory deficits
in AD model mice, such as memantine, and comparing their effects
with those of magnolol might be more rigorous. Second, the
pathological process of AD also involves apoptosis of hippocampal
neurons, and more detailed experimental methods such as Nissl
staining are needed to evaluate neuronal damage. Third, evaluating
the spatial memory of mice through MWM test alone is not
sufficient, and more classic behavioral and memory tests, such as
the open field behavior test, Y-maze test and novel object
recognition test, should be performed to assess the locomotor
ability, short-term spatial memory and recognition memory,
respectively. These experimental methods will be used in future
studies.
In conclusion, the present study reveals a broad
mechanism of action for magnolol as a neuroprotective agent in the
progression of AD. Magnolol exhibited potential in inhibiting Aβ
plaques and neuroinflammation, as well as in reducing the formation
of neurofibrillary tangles, restoring synaptic plasticity, and
inhibiting the activation of astrocytes and microglia. The findings
delineated in the present report could offer crucial data and
perspectives for potential clinical interventions for individuals
suffering from AD.
Acknowledgements
Not applicable.
Funding
Funding: This study was funded by the Science and Technology
Project of Jinhua City of Zhejiang Province in China (grant nos.
2023-4-034, 2023-4-043 and 2023-3-160) and the Science and
Technology Project of Wuyi County of Zhejiang Province in China
(grant no. 2023020).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
LW and MD made substantial contributions to the
conception and design of the study. QY, YS, YW, RL and HZ made
substantial contributions to the acquisition, analysis and
interpretation of the data. QY drafted the manuscript. All authors
critically revised the manuscript for intellectual content. LW, MD,
and QY confirm the authenticity of all the raw data. All authors
have read and approved the final version of the manuscript.
Ethics approval and consent to
participate
This study was approved by The Ethical Committee of
The School of Medicine of Jinhua Polytechnic (Jinhua, China;
approval no. AL-JSYJ202334).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Culig L, Chu X and Bohr VA: Neurogenesis
in aging and age-related neurodegenerative diseases. Ageing Res
Rev. 78(101636)2022.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Joe E and Ringman JM: Cognitive symptoms
of Alzheimer's disease: Clinical management and prevention. BMJ.
367(l6217)2019.PubMed/NCBI View Article : Google Scholar
|
|
3
|
No authors listed. 2023 Alzheimer's
disease facts and figures. Alzheimers Dement. 19:1598–1695.
2023.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Scheltens P, De Strooper B, Kivipelto M,
Holstege H, Chételat G, Teunissen CE, Cummings J and van der Flier
WM: Alzheimer's disease. Lancet. 397:1577–1590. 2021.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Breijyeh Z and Karaman R: Comprehensive
review on Alzheimer's disease: Causes and treatment. Molecules.
25(5789)2020.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Stefanoska K, Gajwani M, Tan ARP, Ahel HI,
Asih PR, Volkerling A, Poljak A and Ittner A: Alzheimer's disease:
Ablating single master site abolishes tau hyperphosphorylation. Sci
Adv. 8(eabl8809)2022.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Mandlik DS, Mandlik SK and S A:
Therapeutic implications of glycogen synthase kinase-3β in
Alzheimer's disease: A novel therapeutic target. Int J Neurosci.
134:603–619. 2024.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Utz J, Berner J, Muñoz LE, Oberstein TJ,
Kornhuber J, Herrmann M, Maler JM and Spitzer P: Cerebrospinal
fluid of patients with Alzheimer's disease contains increased
percentages of synaptophysin-bearing microvesicles. Front Aging
Neurosci. 13(682115)2021.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Gao L, Zhang Y, Sterling K and Song W:
Brain-derived neurotrophic factor in Alzheimer's disease and its
pharmaceutical potential. Transl Neurodegener. 11(4)2022.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Thakur S, Dhapola R, Sarma P, Medhi B and
Reddy DH: Neuroinflammation in Alzheimer's disease: Current
progress in molecular signaling and therapeutics. Inflammation.
46:1–17. 2023.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Li Y, Schindler SE, Bollinger JG, Ovod V,
Mawuenyega KG, Weiner MW, Shaw LM, Masters CL, Fowler CJ,
Trojanowski JQ, et al: Validation of plasma amyloid-β 42/40 for
detecting Alzheimer disease amyloid plaques. Neurology.
98:e688–e699. 2022.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Bloom GS: Amyloid-β and tau: The trigger
and bullet in Alzheimer disease pathogenesis. JAMA Neurol.
71:505–508. 2014.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Selkoe DJ and Hardy J: The amyloid
hypothesis of Alzheimer's disease at 25 years. EMBO Mol Med.
8:595–608. 2016.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Silva MVF, Loures CMG, Alves LCV, de Souza
LC, Borges KBG and Carvalho MDG: Alzheimer's disease: Risk factors
and potentially protective measures. J Biomed Sci.
26(33)2019.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Graham WV, Bonito-Oliva A and Sakmar TP:
Update on Alzheimer's disease therapy and prevention strategies.
Annu Rev Med. 68:413–430. 2017.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Maione F, Piccolo M, De Vita S, Chini MG,
Cristiano C, De Caro C, Lippiello P, Miniaci MC, Santamaria R,
Irace C, et al: Down regulation of pro-inflammatory pathways by
tanshinone IIA and cryptotanshinone in a non-genetic mouse model of
Alzheimer's disease. Pharmacol Res. 129:482–490. 2018.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Ben-Azu B, Nwoke EE, Aderibigbe AO,
Omogbiya IA, Ajayi AM, Olonode ET, Umukoro S and Iwalewa EO:
Possible neuroprotective mechanisms of action involved in the
neurobehavioral property of naringin in mice. Biomed Pharmacother.
109:536–546. 2019.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Jha AB, Panchal SS and Shah A: Ellagic
acid: Insights into its neuroprotective and cognitive enhancement
effects in sporadic Alzheimer's disease. Pharmacol Biochem Behav.
175:33–46. 2018.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Ai-Ping M, Heng-Chang W, Jian-Qiang LI and
Yong-Kang S: A karyomorphological study of 40 species in 11 genera
of the Magnoliaceae from China. J Syst Evol. 44:47–63. 2006.
|
|
20
|
Parray HA, Lone J, Park JP, Choi JW and
Yun JW: Magnolol promotes thermogenesis and attenuates oxidative
stress in 3T3-L1 adipocytes. Nutrition. 50:82–90. 2018.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Chen S, Shen J, Zhao J, Wang J, Shan T, Li
J, Xu M, Chen X, Liu Y and Cao G: Magnolol suppresses pancreatic
cancer development in vivo and in vitro via negatively regulating
TGF-β/Smad signaling. Front Oncol. 10(597672)2020.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Chen Y, Chen S, Chen K, Ji L and Cui S:
Magnolol and 5-fluorouracil synergy inhibition of metastasis of
cervical cancer cells by targeting PI3K/AKT/mTOR and EMT pathways.
Chin Herb Med. 16:94–105. 2023.PubMed/NCBI View Article : Google Scholar
|
|
23
|
He NN, Wang JA, Huang D, Sun XL, Ding F,
Zhao L, Zhang YX, Li HM and Wu CZ: Semisynthesis and in vitro
anti-cancer effect of new magnolol derivatives on the cell
proliferation, apoptosis, migration, and invasion of human
hepatocellular carcinoma SMMC-7721 cells. Chem Pharm Bull (Tokyo).
71:798–803. 2023.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Liu CM, Chen SH, Liao YW, Yu CH, Yu CC and
Hsieh PL: Magnolol ameliorates the accumulation of reactive
oxidative stress and inflammation in diabetic periodontitis. J
Formos Med Assoc. 120:1452–1458. 2021.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Wang X and Jia J: Magnolol improves
Alzheimer's disease-like pathologies and cognitive decline by
promoting autophagy through activation of the AMPK/mTOR/ULK1
pathway. Biomed Pharmacother. 161(114473)2023.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Chen T, Shou L, Guo X, Wei M, Zheng H and
Tao T: Magnolol attenuates the locomotor impairment, cognitive
deficit, and neuroinflammation in Alzheimer's disease mice with
brain insulin resistance via up-regulating miR-200c. Bioengineered.
13:531–543. 2022.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Zhu G, Fang Y, Cui X, Jia R, Kang X and
Zhao R: Magnolol upregulates CHRM1 to attenuate amyloid-β-triggered
neuronal injury through regulating the cAMP/PKA/CREB pathway. J Nat
Med. 76:188–199. 2022.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Talwar P, Sinha J, Grover S, Rawat C,
Kushwaha S, Agarwal R, Taneja V and Kukreti R: Dissecting complex
and multifactorial nature of Alzheimer's disease pathogenesis: A
clinical, genomic, and systems biology perspective. Mol Neurobiol.
53:4833–4864. 2016.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Ferrari C and Sorbi S: The complexity of
Alzheimer's disease: An evolving puzzle. Physiol Rev.
101:1047–1081. 2021.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Calvo-Flores Guzmán B, Elizabeth Chaffey
T, Hansika Palpagama T, Waters S, Boix J, Tate WP, Peppercorn K,
Dragunow M, Waldvogel HJ, Faull RLM and Kwakowsky A: The interplay
between beta-amyloid 1-42 (Aβ1-42)-induced hippocampal
inflammatory response, p-tau, vascular pathology, and their
synergistic contributions to neuronal death and behavioral
deficits. Front Mol Neurosci. 13(522073)2020.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Wang LS, Tao X, Liu XM, Zhou YF, Zhang MD,
Liao YH, Pan RL and Chang Q: Cajaninstilbene acid ameliorates
cognitive impairment induced by intrahippocampal injection of
amyloid-β1-42 oligomers. Front Pharmacol.
10(1084)2019.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Baerends E, Soud K, Folke J, Pedersen AK,
Henmar S, Konrad L, Lycas MD, Mori Y, Pakkenberg B, Woldbye DPD, et
al: Modeling the early stages of Alzheimer's disease by
administering intracerebroventricular injections of human native Aβ
oligomers to rats. Acta Neuropathol Commun. 10(113)2022.PubMed/NCBI View Article : Google Scholar
|
|
33
|
National Research Council (US) Committee
for the Update of the Guide for the Care and Use of Laboratory
Animals: Guide for the care and use of laboratory animals, 8th
edition. Washington (DC): National Academies Press (US), 2011.
|
|
34
|
Rahman SO, Kaundal M, Salman M,
Shrivastava A, Parvez S, Panda BP, Akhter M, Akhtar M and Najmi AK:
Alogliptin reversed hippocampal insulin resistance in an
amyloid-beta fibrils induced animal model of Alzheimer's disease.
Eur J Pharmacol. 889(173522)2020.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Elibol B, Beker M, Terzioglu-Usak S, Dalli
T and Kilic U: Thymoquinone administration ameliorates Alzheimer's
disease-like phenotype by promoting cell survival in the
hippocampus of amyloid beta1-42 infused rat model.
Phytomedicine. 79(153324)2020.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Lana D, Ugolini F, Nosi D, Wenk GL and
Giovannini MG: The emerging role of the interplay among astrocytes,
microglia, and neurons in the hippocampus in health and disease.
Front Aging Neurosci. 13(651973)2021.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Sharma K, Pradhan S, Duffy LK, Yeasmin S,
Bhattarai N and Schulte MK: Role of receptors in relation to
plaques and tangles in Alzheimer's disease pathology. Int J Mol
Sci. 22(12987)2021.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Andrade-Talavera Y and Rodriguez-Moreno A:
Synaptic plasticity and oscillations in Alzheimer's disease: A
complex picture of a multifaceted disease. Front Mol Neurosci.
14(696476)2021.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Deng Q, Wu C, Parker E, Liu TC, Duan R and
Yang L: Microglia and astrocytes in Alzheimer's disease:
Significance and summary of recent advances. Aging Dis.
15:1537–1564. 2024.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Yan T, Liu B, Wang N, Liao Z, Wu B, He B
and Jia Y: The flavonoids of okra insulates against oxidative
stress, neuroinflammation and restores BDNF levels in
Aβ1-42 induced mouse model of Alzheimer's disease. Exp
Gerontol. 147(111263)2021.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Souza LC, Jesse CR, Del Fabbro L, de Gomes
MG, Gomes NS, Filho CB, Goes ATR, Wilhelm EA, Luchese C, Roman SS
and Boeira SP: Aging exacerbates cognitive and anxiety alterations
induced by an intracerebroventricular injection of
amyloid-β1-42 peptide in mice. Mol Cell Neurosci.
88:93–106. 2018.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Giacomeli R, Izoton JC, Dos Santos RB,
Boeira SP, Jesse CR and Haas SE: Neuroprotective effects of
curcumin lipid-core nanocapsules in a model Alzheimer's disease
induced by β-amyloid 1-42 peptide in aged female mice. Brain Res.
1721(146325)2019.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Liu Y, Xu S, Bian H, Qian Y, Li H, Shu S,
Chen J, Cao X, Gu Y, Jin J, et al: Xingnaojing ameliorates synaptic
plasticity and memory deficits in an Aβ1-42 induced
mouse model of Alzheimer's disease. J Pharmacol Sci. 143:245–254.
2020.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Huang W, Li Z, Zhao L and Zhao W:
Simvastatin ameliorate memory deficits and inflammation in clinical
and mouse model of Alzheimer's disease via modulating the
expression of miR-106b. Biomed Pharmacother. 92:46–57.
2017.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Yang Y, Ji WG, Zhang YJ, Zhou LP, Chen H,
Yang N and Zhu ZR: Riluzole ameliorates soluble
Aβ1-42-induced impairments in spatial memory by
modulating the glutamatergic/GABAergic balance in the dentate
gyrus. Prog Neuropsychopharmacol Biol Psychiatry.
108(110077)2021.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Chen Q, Yin Y, Li L, Zhang Y, He W and Shi
Y: Milrinone ameliorates the neuroinflammation and memory function
of Alzheimer's disease in an APP/PS1 mouse model. Neuropsychiatr
Dis Treat. 17:2129–2139. 2021.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Arabi A, Karimi SA, Salehi I, Haddadi R
and Komaki A: Effects of sesamin on Aβ1-42-induced
oxidative stress and LTP impairment in a rat model of Alzheimer's
disease. Metab Brain Dis. 38:1503–1511. 2023.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Twarowski B and Herbet M: Inflammatory
processes in Alzheimer's disease-pathomechanism, diagnosis and
treatment: A review. Int J Mol Sci. 24(6518)2023.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Thal DR and Tomé SO: The central role of
tau in Alzheimer's disease: From neurofibrillary tangle maturation
to the induction of cell death. Brain Res Bull. 190:204–217.
2022.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Sayas CL and Ávila J: GSK-3 and tau: A key
duet in Alzheimer's disease. Cells. 10(721)2021.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Ochs SM, Dorostkar MM, Aramuni G, Schön C,
Filser S, Pöschl J, Kremer A, Van Leuven F, Ovsepian SV and Herms
J: Loss of neuronal GSK3β reduces dendritic spine stability and
attenuates excitatory synaptic transmission via β-catenin. Mol
Psychiatry. 20:482–489. 2015.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Cuestas Torres DM and Cardenas FP:
Synaptic plasticity in Alzheimer's disease and healthy aging. Rev
Neurosci. 31:245–268. 2020.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Liu E, Xie AJ, Zhou Q, Li M, Zhang S, Li
S, Wang W, Wang X, Wang Q and Wang JZ: GSK-3β deletion in dentate
gyrus excitatory neuron impairs synaptic plasticity and memory. Sci
Rep. 7(5781)2017.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Lu B, Nagappan G, Guan X, Nathan PJ and
Wren P: BDNF-based synaptic repair as a disease-modifying strategy
for neurodegenerative diseases. Nat Rev Neurosci. 14:401–416.
2013.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Chen Y and Colonna M: Microglia in
Alzheimer's disease at single-cell level. Are there common patterns
in humans and mice? J Exp Med. 218(e20202717)2021.PubMed/NCBI View Article : Google Scholar
|