Introduction
In breast cancer therapy, the anthracyclines (e.g.,
doxorubicin and daunorubicin) have been widely used either as a
single agent or in the majority of combination protocols (1–3). The
anticancer effects of these agents are mainly through the
inhibition of DNA topoisomerase II (Topo II), which has been
targeted by many clinically important anticancer agents including
doxorubicin and etoposide (4,5).
Although anthracyclines produce significant response rates in
breast cancer patients, their use is hampered by cumulative
dose-limiting cardiotoxicity (6)
and by the development of drug resistance. Therefore, there is an
urgent need to develop novel therapeutic strategies including
alternatives to anthracyclines and/or their combination with other
effective agents for breast cancer treatment.
In an attempt to search for new alternatives to
anthracyclines which are less toxic and less prone to elicit
resistance, the compound
2-({4-[4-(acridin-9-ylamino)phenylthio]phenyl}
(2-hydroxyethyl)amino)ethan-1-ol (CK0402) was selected as a
potential anticancer agent among the series of sulfur-containing
9-aminoacridine analogues previously synthesized by our group for
treatment against breast cancer (7). CK0402 is a DNA intercalating Topo II
inhibitor and has been shown to exert a growth inhibition effect
against various types of cancer cell lines; the anticancer activity
of CK0402 was also demonstrated in vivo in a study in which
it significantly increased the lifespan of P388 leukemia-bearing
mice (7,8). Unlike anthracyclines, CK0402 is not
structurally related to quinone which has been responsible for the
generation of free radicals in many active analogues and their
mediated toxicity. It was also proposed that the structural
modification of 9-aminoacridine analogues as in CK0402 may overcome
the multidrug resistance induced by doxorubicin and etoposide
(9). Thus, CK0402 has the
potential to serve as a safer and more efficient alternative to
anthracyclines.
In this study, we examined the cytotoxic effects of
CK0402 in a panel of established human breast cancer cell lines
with varying levels of the estrogen receptor (ER) and
HER2/neu, which are the two most common clinically used
biomarkers for breast cancer treatment. These cell lines include
MCF-7 [ER(+) and HER2(−)], BT-474 [ER(+) and HER2-overexpressing],
T-47D [ER(+) and HER2-expressing], MDA-MB-231 [ER(−) and HER2(−)]
and SKBR-3 [ER(−) and HER2-overexpressing]. To elucidate the
mechanism underlying the growth inhibition activity of CK0402 in
breast cancer cells, the ability of CK0402 to alter cell cycle
progression and the nature of the cell death response induced by
CK0402 were also examined. Combination strategy has been widely
used to enhance the efficacy of chemotherapy. Trastuzumab
(Herceptin®), a humanized monoclonal antibody targeting
the extracellular domain of the tyrosine kinase receptor HER2, has
shown additive/synergistic anticancer activity with
chemotherapeutic agents including anthracyclines in
HER2-overexpressed metastatic breast cancer (10–12).
Overexpression of the HER2/neu gene has been found in 20–25%
of patients with breast cancer and is correlated with higher stages
of malignancy (13). Since SKBR-3
cells are HER2/neu-overexpressing, Topo II amplifying cells
and sensitive to Herceptin treatment, the SKBR-3 cell line was
chosen to investigate the combination effect of CK0402 and
Herceptin.
Materials and methods
Chemicals, reagents and cell lines
CK0402 was synthesized and purified as described
previously (7). Herceptin was
kindly provided by Genentech Inc. (South San Francisco, CA).
Dimethyl sulfoxide (DMSO, cell culture grade), chloroquine,
sulforhodamine B and propidium iodide were purchased from
Sigma-Aldrich Chemical Co. (St. Louis, MO). Dulbecco’s modified
Eagle’s medium (DMEM)/F12, Iscove’s modified Dulbecco’s medium
(IMDM), penicillin/streptomycin, phosphate-buffered saline (PBS),
RNase A and trypsin-EDTA were purchased from Gibco-Invitrogen.
Fetal bovine serum (FBS) was purchased from Gemini Bio-Products
(Woodland, CA). MCF-7, BT-474, T-47D, MDA-MB-231 and SKBR-3 cells
were obtained from the American Type Culture Collection (Manassas,
VA).
Cell culture and treatment
MCF-7 and MDA-MB-231 cells were maintained in
IMDM/F12 (1:1 mixture) medium; SKBR-3, T47-D and BT-474 cells were
maintained in DMEM/F12 (1:1 mixture). All the media were
supplemented with 10% FBS and penicillin/streptomycin (50 μg/ml).
Cells were grown from frozen stock and maintained at 37°C in a
humidified atmosphere with 5% CO2. Drug treatment
involved continuous exposure to the compound. For all cell culture
experiments, cells were allowed to seed at least 24 h before
treatment.
Assessment of viable cell number
Cell viability was determined by trypan blue
exclusion at various time points after the initiation of drug
treatment. Cells were harvested by trypsinization, stained with
0.4% trypan blue dye and counted using phase contrast microscopy on
a hemacytometer. Cells that excluded trypan blue dye were
considered to be viable.
Cell proliferation assay and multiple
drug effect analyses
Cells (approximately 3,000–5,000/well) were seeded
and grown in a 96-well plate for at least 24 h before treatment.
CK0402 was dissolved in DMSO, yielding a final DMSO concentration
≤0.1% (v/v) in the medium. When cells were treated with the
combination of CK0402 and Herceptin, Herceptin was added 3 h prior
to the addition of CK0402. At the end of the incubation, cultures
were fixed with 50 μl of 50% cold trichloroacetic acid and
incubated at 4°C for 1 h. The plates were washed five times with
water and then air-dried. The fixed cells were stained for 30 min
with 100 μl of 0.4% sulforhodamine B solution in 1% acetic acid. At
the end of staining, the plate was washed five times with 1% acetic
acid to remove unbound dye. The bound dye was dissolved with 10 mM
Tris buffer, and the absorbance of the resulting solution was
measured at 570 nm to quantify the number of surviving cells. All
treatments were in triplicate and performed at least three times.
The drug concentration that produced a 50% reduction
(LC50) in cell survival was determined by the
median-effect plot. Combined effects of CK0402 and Herceptin in
SKBR-3 cells were analyzed by the multiple drug effect analysis of
Chou and Talalay (14).
LC50 and the combination index (CI) were calculated by
the program CompuSyn (CompuSyn, Paramus, NJ). CI values were
derived from variables of the median effect plots, and statistical
tests (Student’s t-test) were applied to determine whether the mean
CI values at multiple effect levels were significantly different
from CI=1. In this method, synergy is defined as CI values
significantly <1.0, additivity as CI values =1.0 and antagonism
as CI values significantly >1.0.
Flow cytometry of cell cycle
analysis
MCF-7 and SKBR-3 cells were grown to exponential
phase and treated with the indicated concentration of CK0402 or
DMSO. At the end of the incubation, cells were harvested, fixed in
ice-cold 70% ethanol and stored at −20°C. The fixed cells were
washed with phosphate-buffered saline, treated with RNase A (3
U/ml) at 37°C for 30 min, and stained with propidium iodide (50
μg/ml) for 5 min. DNA content for 250,000 cells per analysis was
monitored with a Becton-Dickinson FACScan flow cytometer, and
Modfit software (LT version 2.0) was used for the analysis of cell
cycle status.
Apoptotic cell death detection
A cell death detection enzyme-linked immunosorbent
assay kit (Roche Diagnostics, Indianapolis, IN) was used to
quantitatively determine cytoplasmic histone-associated DNA
oligonucleosome fragments associated with apoptotic cell death,
according to the manufacturer’s manual. Briefly, after cells were
lysed and incubated for 30 min at room temperature, 20 μl of
supernatant was transferred into the streptavidin-coated microtiter
plate, and 80 μl of the immunoreagent was added to each well. After
incubation at room temperature for 2 h, the solution was decanted,
and each well was rinsed three times with incubation buffer. Color
development was carried out by adding 100 μl of ABTS solution, and
absorbency was measured at 405 nm in a microtiter plate reader
against ABTS solution as a blank.
Western blot analysis
Cells treated with various doses of CK0402 at
various incubation times were harvested by scraping and then washed
with PBS. Cellular proteins were isolated with cell lysis buffer
purchased from Cell Signaling Technology (Beverly, MA). Equal
amount of protein (40 μg) was taken, boiled for 5 min,
electrophoresed on a 10% SDS-PAGE at 100 V for 110 min, then
electro-transferred to PVDF membranes. Antibody against LC-3 was
purchased from NanoTools (Munich, Germany). Antibody against
caspase-3, cleaved caspase-3 and caspase-7 were obtained from Cell
Signaling Technology. Mouse monoclonal antibody against α-tubulin
was purchased from Sigma-Aldrich Chemical Co. After extensive
washing, specific bands were detected using Immobilon Western
Chemiluminescent substrate (Millipore, MA). Secondary anti-mouse or
anti-rabbit IgG, conjugated with horseradish peroxidase (HRP), were
purchased from Cell Signaling Technology.
Results
Growth inhibition activity of CK0402 in
human breast cancer cells
The effects of CK0402 on human breast cancer cells
were evaluated in a panel of established human breast cancer cell
lines which express varied levels of ER and HER2. Initial studies
were conducted to examine the time-dependent growth inhibition
effects of CK0402 in MCF-7 [ER(+) and HER2(−)] and SKBR-3 cells
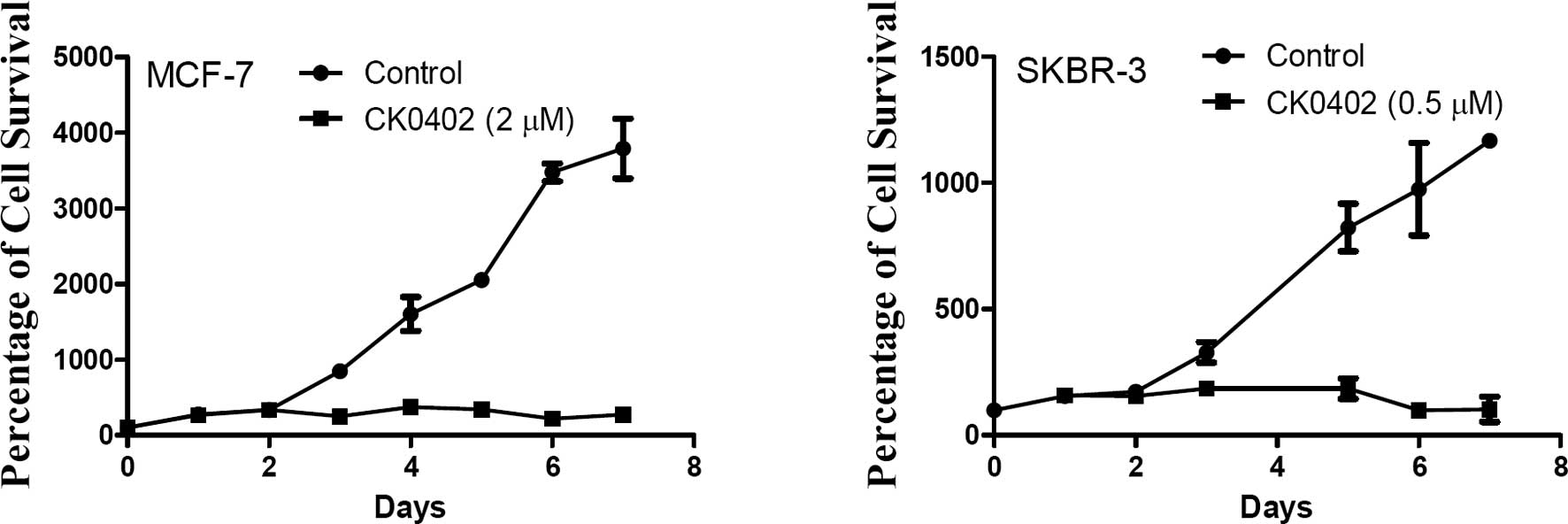
[ER(−) and HER2-overexpressing]. Fig.
1A and B shows the time-dependent growth inhibition effect of
CK0402 in MCF-7 and SKBR-3 cells, respectively. Although no direct
cell killing effect of CK0402 was observed within the 48 h of drug
exposure, the growth inhibition effect was consistently observed
after 72 h in both cell lines. Based on our observations, we chose
to determine the LC50 of CK0402 in the selected cell
lines after 6 days of continuous drug exposure. Subsequently,
sulforhodamine B protein assay was used to estimate cell viability,
and LC50 was calculated. With this well-established
methodology, the growth inhibitory activity of CK0402 was
demonstrated in all of the cell lines, and values of
LC50 were determined (0.29–3.07 μM) (Table I). Of the cell lines tested, the
ER(−) and HER2-overexpressing SKBR-3 cell line was the most
sensitive (LC50=0.29), while the ER(+) and
HER2-overexpressing BT-474 cell line was the most insensitive to
CK0402 treatment. Although both SKBR-3 and MDA-MB-231 cells are
ER(−) and appeared to be more sensitive to CK0402 than ER(+) MCF-7,
T-47D and BT-474 cells, correlations between hormone receptor
status and sensitivity to CK0402 would be speculative and could not
be established in this study (15).
 | Table I.LC50 of CK0402 in a panel
of breast cancer cells. |
Table I.
LC50 of CK0402 in a panel
of breast cancer cells.
| Cell lines | LC50
(μM)a |
|---|
| MCF-7 | 0.65±0.21 |
| MDA-MB-231 | 0.44±0.02 |
| BT-474 | 3.07±1.72 |
| T-47D | 0.97±0.18 |
| SKBR-3 | 0.29±0.03 |
Cell cycle analysis
To investigate the mechanisms underlying the growth
inhibition activity of CK0402, we examined the cell cycle
distribution in both MCF-7 [ER(+) and HER2(−)] and SKBR-3 [ER(−)
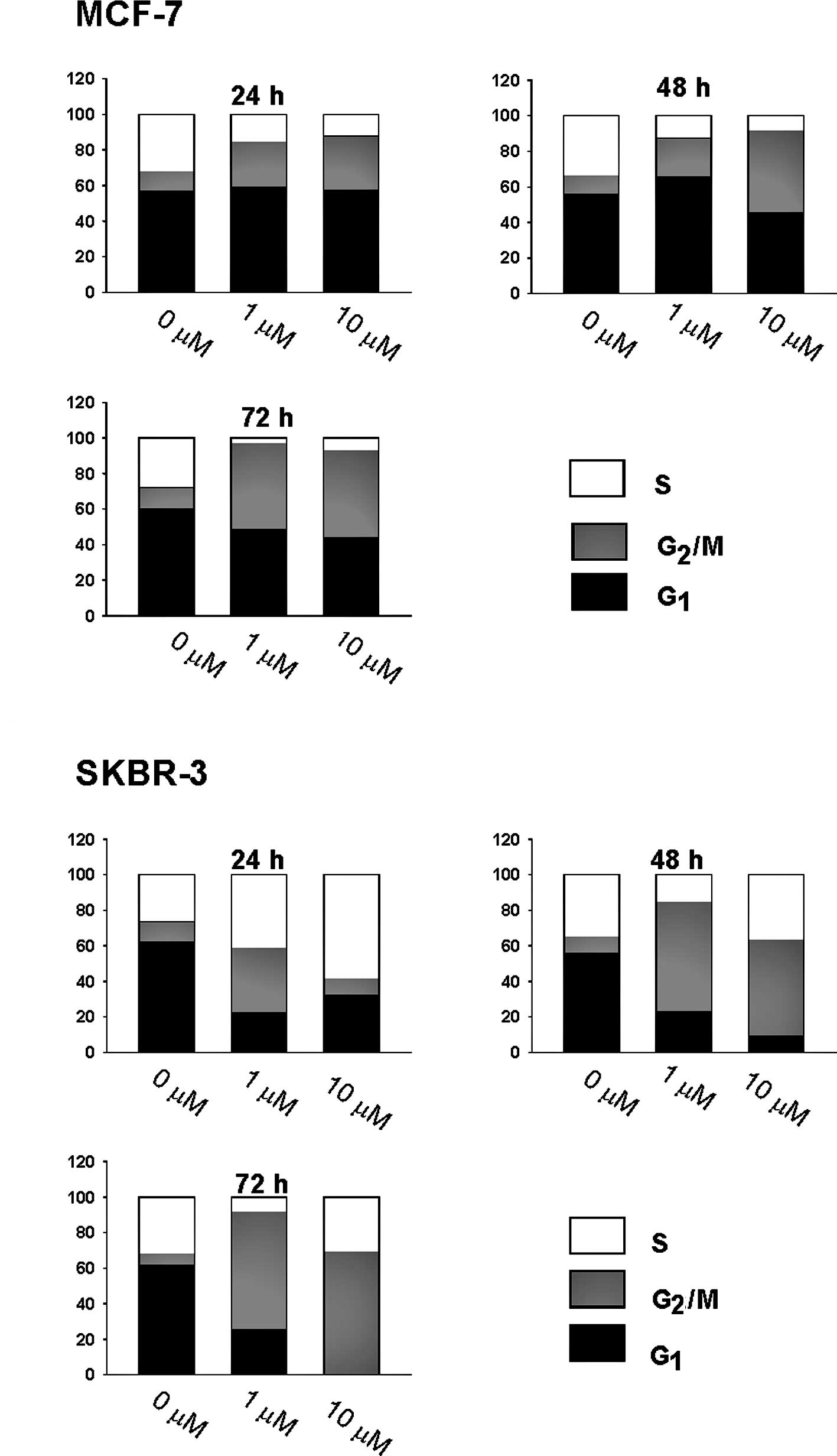
and HER2-overexpressing] cells treated with CK0402. We found that
after continuous exposure of cells to 1 and 10 μM of CK0402 for 24,
48 and 72 h, CK0402 induced concentration- and time-dependent
G2/M arrest with increased G2/S ratio in
MCF-7 cells (Fig. 2A). However,
the same treatment in SKBR-3 cells induced a transient S phase
accumulation at 24 h, but cells progressed to accumulate in the
G2/M phase by 72 h with a massive loss of cells in the
G1 phase (Fig. 2B). The
lack of S phase cell accumulation in MCF-7 cells was also confirmed
by conducting experiments at early time points (data not shown).
Our results showed that the induction of G2/M arrest by
CK0402 was weaker in MCF-7 cells than in SKBR-3 cells; in addition,
G2/M arrest in MCF-7 cells was not accompanied by a
massive reduction of cells in the G1 phase. In fact, a
substantial fraction of MCF-7 cells remained in the G1
phase even after 6 days of exposure to CK0402 (data not shown).
Induction of apoptosis and autophagy
To characterize the cell death response induced by
CK0402, analysis of apoptosis was performed in both MCF-7 and
SKBR-3 cells using an enzyme-linked immunosorbent ELISA kit, which
quantitatively determines the cytoplasmic histone-associated DNA
oligonucleosome fragments associated with apoptotic cell death. An
initial study was conducted at 24, 48 and 72 h in MCF-7 cells
treated with various concentrations of CK0402. However, an
apoptotic effect was not observed at 24 and 48 h (data not shown).
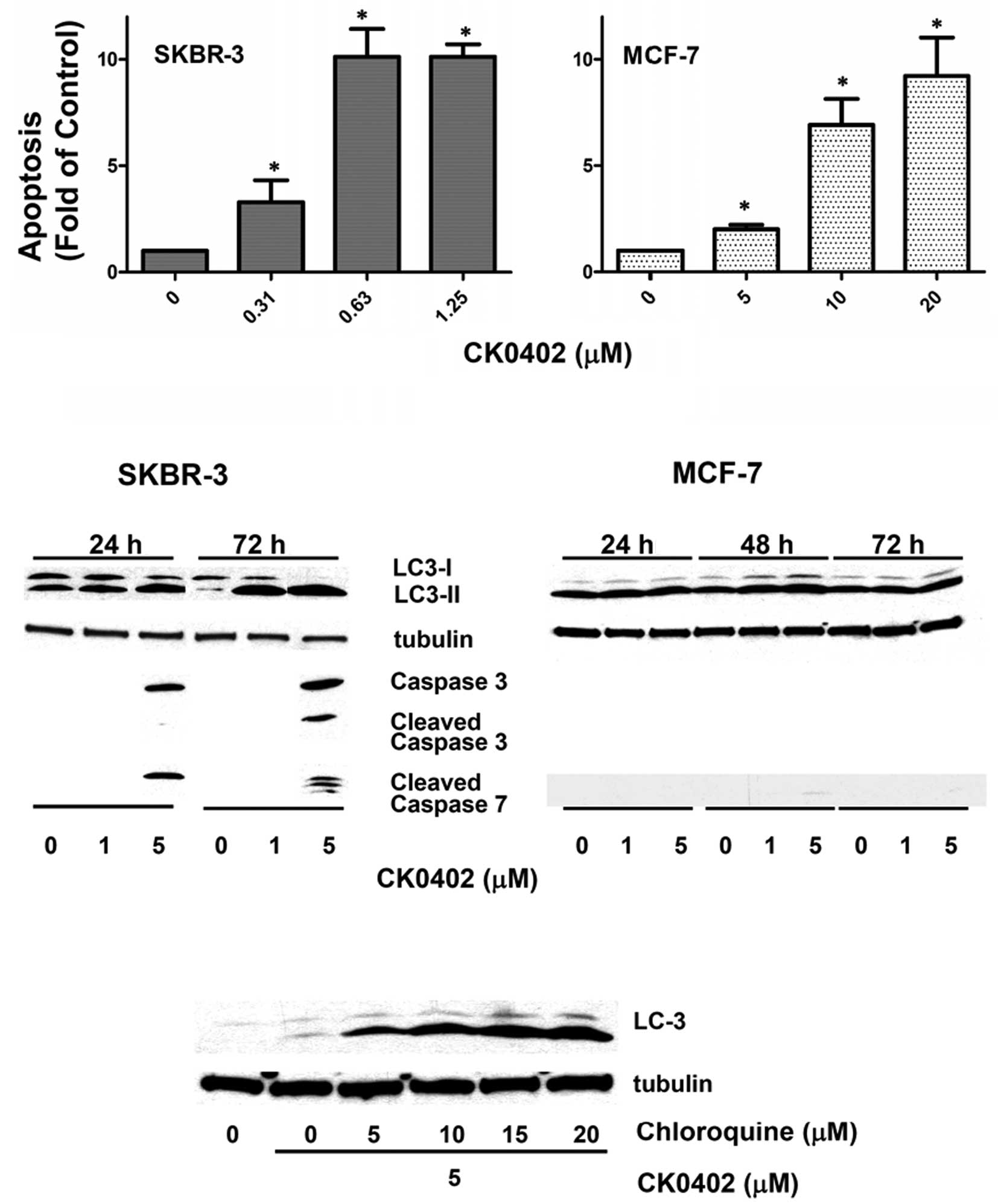
At 72 h, a concentration-dependent apoptosis induced by CK0402 in
MCF-7 cells (doses 5, 10 and 20 μM) was observed, but the apoptotic
effect in MCF-7 cells was not as potent as that observed in SKBR-3
cells (doses 0.31, 0.63 and 1.25 μM) (Fig. 3A). To further investigate the
apoptotic events involved in CK0402-induced apoptosis, we examined
the levels of caspase proteins in SKBR-3 and MCF-7 cells by Western
blot analysis. In the case of SKBR-3 cells, which exhibited a
strong apoptosis response in the ELISA assay at a dose as low as
0.63 μM, activation of caspase-3/7 was only observed at a dose of 5
μM (Fig. 3B), but not at a dose of
1 μM. In MCF-7, which is a caspase-3 deficient cell line, no
caspase-7 activation was detected at 24 and 72 h. These results
suggest that other caspase-independent pathways may be involved in
the cell death process induced by CK0402.
It is known that therapeutic stresses activate
pathways that regulate both apoptosis and autophagy. We next
examined the effect of CK0402 on the induction of autophagy in
MCF-7 and SKBR-3 cells. Autophagy was evaluated by detecting
microtubule-associated protein 1 light chain 3-II (LC3-II)
expression using Western blot analysis. LC3 is a known protein that
specifically associates with autophagosomes. The antibody-based
detection of LC3 expression is the ‘gold standard’ for molecularly
defined detection of autophagy, and the processing from LC3-I to
LC3-II is required for the formation of autophagosome (16,17).
Our results showed that treatment with CK0402 in SKBR-3 cells
induced LC3 activation and cleavage in a dose- and time-dependent
manner (Fig. 3B). However, CK0402
exhibited a weak effect on LC3 activation in MCF-7 cells. The
induction of autophagy by CK0402 in SKBR-3 cells was further
confirmed by the addition of chloroquine, which enhanced the
accumulation of LC3-II in a dose-dependent manner (Fig. 3C). Although the role of autophagy
in the cytotoxic effect of CK0402 remains to be identified, these
results clearly showed that CK0402 induced autophagy in breast
cancer SKBR-3 cells.
Multiple drug effect analysis
HER2-overexpressing SKBR-3 cells were the most
sensitive cells to CK0402 treatment and were selected to evaluate
the combination effect of CK0402 and the HER2-targeted Herceptin.
Parallel experiments were also conducted with HER2(−) MCF-7 cells
as a negative control. CI values for the combination of CK0402 and
Herceptin at drug ratios (CK0402/Herceptin) from 1 to 200 at the
LC50 are summarized in Table II. Results showed that in SKBR-3
cells, CK0402 exhibited synergistic interaction (CI<1;
P<0.001) with Herceptin at the drug ratios ranging from 10 to
200, while an additive interaction was found at a drug ratio equal
to 1. No synergistic or additive effect was observed in MCF-7 cells
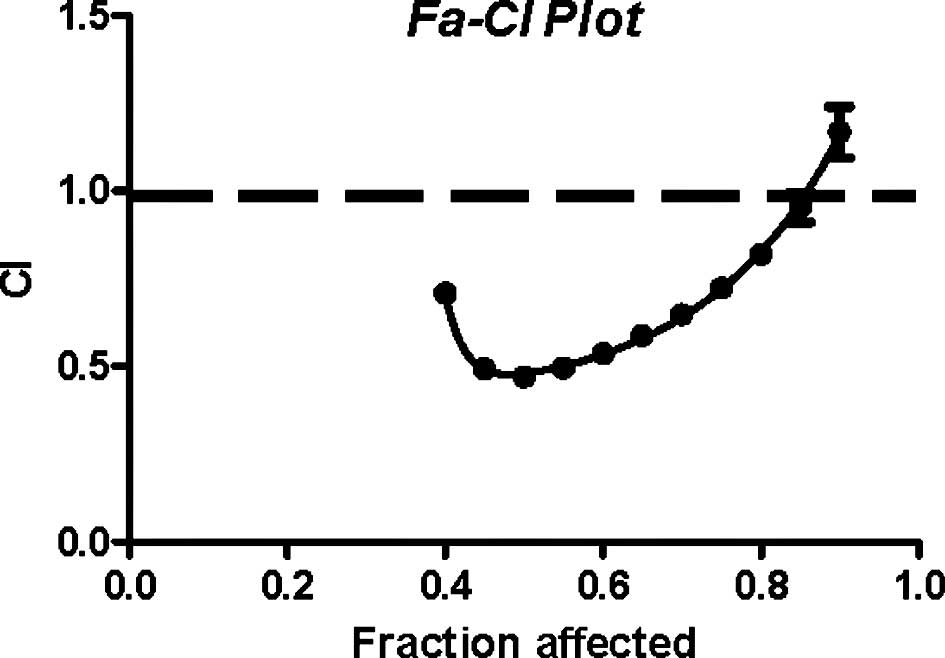
(data not shown). The lowest CI value (0.47) was observed at a drug
ratio 10; therefore, a Fa-CI plot (Fig. 4) of the fraction of affected cells
(Fa) vs. CI was constructed at the drug ratio 10. This plot
indicated synergism at the range from LC40 to
LC85 (P<0.007), while it indicates an additive effect
at LC90.
| Table II.Combined effects of CK0402 and
Herceptin at various molar ratios in SKBR-3 cells. |
Table II.
Combined effects of CK0402 and
Herceptin at various molar ratios in SKBR-3 cells.
| CK0402/Herceptin
molar ratio | Combination index
(CI) (mean ± SD)a | P-valueb | Interaction |
|---|
| 1 | 1.08±0.09 | 0.13000 | Addition |
| 10 | 0.47±0.02 | <0.00010 | Synergy |
| 100 | 0.69±0.05 | <0.00010 | Synergy |
| 200 | 0.91±0.02 | 0.00026 | Synergy |
Discussion
In the present study, we investigated the
antiproliferative activity of CK0402 in a panel of established
human breast cancer cells including MCF-7, T-47D, BT-474,
MDA-MB-231 and SKBR-3, as well as its combined effect with
Herceptin in SKBR-3 cells. CK0402 is a 9-aminoacridine analogue
which has been shown to inhibit Topo IIα-catalyzed decatenation
reaction (8). There are two highly
related Topo II enzymes coded by different genes: Topo IIα (170
kDa) and Topo IIβ (180 kDa) (18).
Studies have shown that, in general, there is no significant
variation in the expression of Topo IIβ protein levels in breast
cancer cell lines; however, relatively large variations in Topo IIα
protein levels are observed (19).
The expression of Topo IIα protein levels in these cells was
reported as follows: SKBR-3 > MCF-7 > MDA-MB-231 > T-47D
> BT-474 (19,20). The order of LC50 of
these breast cancer cells to CK0402 was determined as SKBR-3 <
MDA-MB-231 < MCF-7 < T-47D < BT-474. Despite the
controversy between Topo IIα expression and response to Topo II
inhibitors (21,22), our results support that Topo IIα
expression was the most possible factor determining the cellular
sensitivity to CK0402 in our panel of breast cancer cells. BT-474
cells, which were found to exert considerable resistance to various
types of chemotherapeutic agents including Topo II inhibitors in
human breast cancer cells (19),
expressed the lowest level of Topo IIα as well as the most
resistance to CK0402 among the tested cells. The two most sensitive
cell lines tested were SKBR-3 and MDA-MB-231 which are both ER(−);
the former is HER2 overexpressed and the latter is not.
The inhibition of Topo II by CK0402 may cause DNA
damage, which may activate pathways that lead to cell cycle arrest
or apoptosis. The former may be a major cellular response to DNA
damage preceding repair or death. G2/M arrest is a
common response after treatment with DNA damaging agents such as
Topo II inhibitors (e.g., doxorubicin, amsacrine and DACA)
(23). The fact that CK0402
induced prolonged G2/M arrest with delayed growth
inhibition in both MCF-7 and SKBR-3 cells suggests that these
slowly dying cells may initiate cell death only after the
accumulation of cells in the G2/M phase, and differences
in the extent of G2/M arrest may be critical
determinants of cellular sensitivity to CK0402.
DNA damage induced by chemotherapeutics may cause
various types of cell death in tumor cells, most likely depending
on the mechanism of drug action, the dosing regimen used and the
type of tumor cells. Although apoptosis has been commonly regarded
as the prevailing mechanism of cell death induced by
chemotherapeutics, it has recently been acknowledged that multiple
modes of cell death combine to generate the overall tumor response
(24). Autophagy is involved in a
variety of cellular functions, including development, response to
nutrient deprivation and cell death (25). Most of the cellular systems in
which autophagy contributes to cell death have involved defects in
the apoptosis signaling pathways, suggesting that these two forms
of cell death can act as backup mechanisms for each other under
conditions where cell death is imperative. However, tumor cells may
undergo, separately or simultaneously, both apoptosis and autophagy
in response to certain chemotherapeutics (26,27).
In this study, the delayed cell death response to CK0402 in MCF-7
cells was characterized by prolonged cell arrest without apparent
apoptosis or autophagy. In SKBR-3 cells, although apoptosis was
observed, the roles of autophagy in the mechanism of cell death
induced by CK0402 need to be further investigated. To the best of
our knowledge, this study is the first to demonstrate that
9-aminoacridine compounds induce autophagy.
The development of anti-HER2 therapies such as
Herceptin (28) has markedly
improved the therapeutic outcomes of adjuvant chemotherapy for
patients with breast cancers that overexpress HER-2 (12). The combination of a Topo II
inhibitor with Herceptin in breast cancer therapy was initially
supported by in vitro studies using Herceptin in combination
with doxorubicin which demonstrated an additive effect, and with
etoposide which showed a synergistic effect. Recent clinical
studies have shown that patients with co-amplification of genes
encoded for Topo IIα and HER2 received a therapeutic advantage from
the anthracycline-Herceptin combination regimens (10,11).
The combination of Herceptin with drugs targeting Topo II was
further justified based on the findings that genes encoded for Topo
IIα and HER2 are both located on chromosome 17q and are frequently
co-amplified or deleted (10,11).
In the present study, data from the multiple drug effect analysis
methodology demonstrated that the combination of CK0402 and
Herceptin exerted a maximal synergistic effect at a molar ratio
equal to 10, and the effect was decreased as the molar ratio was
decreased to 1 or increased to 100 and 200 (Table II). Our results demonstrate that
CK0402 possesses a broad spectrum of activity in human breast
cancer cells, and the combination of CK0402 and Herceptin exerts a
synergistic/additive effect in HER2-overexpressing and Topo
IIα-amplified SKBR-3 cells. In conclusion, the synergistic or
additive interaction between CK0402 and Herceptin in SKBR-3 cells
suggests that such a combination may be a potential alternative to
anthracyclines in combination with Herceptin against the more
aggressive ER(–) and HER2-overexpressing breast cancer.
Acknowledgements
This study was supported by the Susan
G. Komen Breast Cancer Foundation (grant no. BCTR0707876), and the
Penn State College of Medicine Dean’s Feasibility Grant.
References
|
1.
|
Weiss RB: The anthracyclines: Will we ever
find a better doxorubicin? Semin Oncol. 19:670–686. 1992.PubMed/NCBI
|
|
2.
|
Gianni L and Valagussa P: Anthracyclines
and early breast cancer: the end of an era? J Clin Oncol.
27:1155–1157. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
O’Shaughnessy J: Liposomal anthracyclines
for breast cancer: overview. Oncologist. 8(Suppl 2): 1–2. 2003.
|
|
4.
|
Liu LF: DNA topoisomerase poisons as
antitumor drugs. Annu Rev Biochem. 58:351–375. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Nitiss JL: Targeting DNA topoisomerase II
in cancer chemotherapy. Nat Rev Cancer. 9:338–350. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Shan K, Lincoff AM and Young JB:
Anthracycline-induced cardiotoxicity. Ann Intern Med. 125:47–58.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Chen KM, Sun YW, Tang YW, Sun ZY and Kwon
CH: Synthesis and antitumor activity of sulfur-containing
9-anilinoacridines. Mol Pharm. 2:118–128. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Park SK, Kang H and Kwon CH:
Caspase-dependent cell death mediates potent cytotoxicity of
sulfide derivatives of 9-anilinoacridine. Anticancer Drugs.
19:381–389. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Baguley BC, Holdaway KM and Fray LM:
Design of DNA intercalators to overcome topoisomerase II-mediated
multidrug resistance. J Natl Cancer Inst. 82:398–402. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Jarvinen TA and Liu ET: Simultaneous
amplification of HER-2 (ERBB2) and topoisomerase IIalpha (TOP2A)
genes – molecular basis for combination chemotherapy in cancer.
Curr Cancer Drug Targets. 6:579–602. 2006.
|
|
11.
|
Jorgensen JT, Nielsen KV and Ejlertsen B:
Pharmacodiagnostics and targeted therapies – a rational approach
for individualizing medical anticancer therapy in breast cancer.
Oncologist. 12:397–405. 2007.
|
|
12.
|
Smith I and Chua S: Medical treatment of
early breast cancer. IV: neoadjuvant treatment. BMJ. 332:223–224.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Nahta R and Esteva FJ: Herceptin:
mechanisms of action and resistance. Cancer Lett. 232:123–138.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Chou TC and Talalay P: Quantitative
analysis of dose-effect relationships: the combined effects of
multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 22:27–55.
1984. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Smith K, Houlbrook S, Greenall M,
Carmichael J and Harris AL: Topoisomerase II alpha co-amplification
with erbB2 in human primary breast cancer and breast cancer cell
lines: relationship to m-AMSA and mitoxantrone sensitivity.
Oncogene. 8:933–938. 1993.PubMed/NCBI
|
|
16.
|
Amaravadi RK and Thompson CB: The roles of
therapy-induced autophagy and necrosis in cancer treatment. Clin
Cancer Res. 13:7271–7279. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Klionsky DJ, Abeliovich H, Agostinis P, et
al: Guidelines for the use and interpretation of assays for
monitoring autophagy in higher eukaryotes. Autophagy. 4:151–175.
2008. View Article : Google Scholar
|
|
18.
|
Baldwin EL and Osheroff N: Etoposide,
topoisomerase II and cancer. Curr Med Chem Anticancer Agents.
5:363–372. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Davis PL, Shaiu WL, Scott GL, Iglehart JD,
Hsieh TS and Marks JR: Complex response of breast epithelial cell
lines to topoisomerase inhibitors. Anticancer Res. 18:2919–2932.
1998.PubMed/NCBI
|
|
20.
|
Houlbrook S, Addison CM, Davies SL, et al:
Relationship between expression of topoisomerase II isoforms and
intrinsic sensitivity to topoisomerase II inhibitors in breast
cancer cell lines. Br J Cancer. 72:1454–1461. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Mano MS, Rosa DD, De Azambuja E, Ismael GF
and Durbecq V: The 17q12–q21 amplicon: Her2 and
topoisomerase-IIalpha and their importance to the biology of solid
tumours. Cancer Treat Rev. 33:64–77. 2007.
|
|
22.
|
Pritchard KI, Messersmith H, Elavathil L,
Trudeau M, O’Malley F and Dhesy-Thind B: HER-2 and topoisomerase II
as predictors of response to chemotherapy. J Clin Oncol.
26:736–744. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Haldane A, Holdaway KM, Finlay GJ and
Baguley BC: Cytokinetic differences in the action of
N-[2-(dimethylamino) ethyl]acridine-4-carboxamide as compared with
that of amsacrine and doxorubicin. Cancer Chemother Pharmacol.
32:463–470. 1993.
|
|
24.
|
Morse DL, Gray H, Payne CM and Gillies RJ:
Docetaxel induces cell death through mitotic catastrophe in human
breast cancer cells. Mol Cancer Ther. 4:1495–1504. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Bursch W, Ellinger A, Gerner C, Frohwein U
and Schulte-Hermann R: Programmed cell death (PCD). Apoptosis,
autophagic PCD, or others? Ann NY Acad Sci. 926:1–12. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Hoyer-Hansen M and Jaattela M: Autophagy:
an emerging target for cancer therapy. Autophagy. 4:574–580. 2008.
View Article : Google Scholar
|
|
27.
|
Yu L, Wan F, Dutta S, et al: Autophagic
programmed cell death by selective catalase degradation. Proc Natl
Acad Sci USA. 103:4952–4957. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Ma WW and Adjei AA: Novel agents on the
horizon for cancer therapy. CA Cancer J Clin. 59:111–137. 2009.
View Article : Google Scholar : PubMed/NCBI
|