Introduction
Breast cancer is the leading cause of morbidity and
mortality in women worldwide. Some types of breast cancer are under
estrogen control (1). By
associating with nuclear estrogen receptors (ERs), estradiol and
selective modulators of estrogen receptors (SMER) affect the growth
of mammary gland neoplasias through the regulation of genomic
processes (2). In addition,
estradiol and certain SMER associate with cognate receptors in the
cell membrane or the cytoplasm, and control non-genomic effects
(3,4).
The concentration of estrogens in breast tissue
depends on the uptake from circulation and local production; the
latter seems to be precisely controlled by the activity of a
variety of nuclear receptors (5).
Local levels of estradiol maintain the operation of numerous
biological functions in normal and neoplasic breast tissue. Of
particular importance among these is cell proliferation. For this
reason, the blockade of ER activation by anti-estrogens and the
reduction in estrogen availability through the use of inhibitors of
aromatase and other enzymes involved in the local synthesis of the
steroid are important therapeutic strategies used to hinder the
progress of ER-positive mammary cancer (6). However, not all ER-positive breast
cancers respond to these treatments, and many tumors eventually
acquire resistance during therapy, probably due to the sustained
functioning of ER-related pathways (7). Therefore, a better understanding of
the mechanisms that maintain the activation of the ER signaling
pathways in cancer cell progression is required for the development
of new therapeutic approaches.
The association of a particular SMER to an ER has an
effect on the overall conformation of the protein. The specific
conformation of the complex determines the differential recruitment
of co-activators or co-repressors as well as the biochemical and
physiological activities to be expressed (7–9).
Serum and the fluids bathing cells, organs and tissues contain
compounds with the potential to imitate estrogens. In this regard,
it has been reported that 27-hydroxycholesterol (27OHC), an
oxysterol produced by the intramitochondrial oxidation of
cholesterol, may act as a non-conventional SMER in estradiol-target
cells (10,11). In normal subjects, plasma 27OHC
concentration is in the same order of magnitude as the dissociation
constant of the complex between ER and 27OHC, thus assuring their
stable association. In endothelial cells, 27OHC functions as an
estradiol antagonist, and some authors assume that its increased
release by atheroma cells is responsible for damage to vascular
endothelial and muscular cells in the vicinity of the
atherosclerotic plaques (10,11).
However, in ER-positive mammary tumor cells 27OHC works as an
estradiol agonist, promoting cell proliferation (11).
In the present study, we compared the activity of
27OHC and estradiol on the proliferation of mammary cancer cells in
culture, and determined their inhibition by the specific antagonist
ICI 182,780. Analysis of the effects of 27OHC on the cell cycle was
also conducted. Additionally, we evaluated the effects of
simvastatin, a specific inhibitor of
3-hydroxy-3-methyl-glutaryl-CoA reductase (HMGR), on the
proliferative activity of 27OHC. Lastly, we studied the effects of
an α-fetoprotein (AFP)-derived cyclopeptide on tumor cell
proliferation under estradiol and 27OHC stimulation.
Materials and methods
Tissue culture materials were obtained from
NalgeNunc (Rochester, NY, USA). 27-hydroxycholesterol (C6570-000)
was purchased from Steraloids Inc. (Newport, RI, USA). Dulbecco’s
phosphate buffered saline (DPBS) was from Gibco-Invitrogen Corp.
(Carlsbad, CA, USA). Pure antiestrogen ICI 182,780 (ICI) was
purchased from Tocris Bioscience (Ellisville, MO, USA). The
majority of the other reagents were purchased from Sigma-Aldrich
Inc. (St. Louis, MO, USA).
The AFP-derived nonapeptide [AFPep,
cyclo(EKTOVNOGN), where O is hydroxyproline] was generously
supplied by Professor H.J. Jacobson and colleages of the Albany
Medical College, NY, USA.
Rabbit anti-c-erbB2 polyclonal antibody (ab2428) was
obtained from Abcam plc (Cambridge, UK). Alexa Fluor 488 conjugated
goat anti-rabbit IgG (A11008) was purchased from Molecular
Probes-Invitrogen Corp. (Carlsbad, CA, USA).
E2-sensitive MCF7 epithelial cells from
human metastatic breast cancer tissue (HTB 22; ATCC, USA) were
cultured in DMEM/F12 containing 10% fetal bovine serum, 1 mM sodium
pyruvate, 2 mM L-glutamine, 100 U/ml penicillin and 100 μg/ml
streptomycin. In the proliferation studies, the cells were
transferred 24 h after seeding to DMEM/F12 containing ITS (insulin,
transferrin and selenium), 1% charcoal/dextran-twice-treated serum
(CDTS), 3% hydroxyethylated starch (HAES), 50 U/ml penicillin and
50 μg/ml streptomycin. The non-tumorigenic
E2-insensitive epithelial cell line MCF10 (CRL-10317;
ATCC, USA) was used for control studies, and was cultured under the
same conditions as MCF7. In each of the experiments, the cells were
incubated at 37°C in a humidified incubator under a 5%
CO2 atmosphere.
Proliferation studies
As indicated above, MCF7 or MCF10 (∼20,000
cells/cm2), were seeded and incubated for 24 h to allow
attachment, then non-adherent cells and media were removed. The
remaining cells were washed and further incubated for various
periods with low-serum culture medium containing either 2 nM
E2 or different concentrations of 27OHC or cholesterol,
in the absence or presence of either 2 μg/ml AFPep, 1 μM
simvastatin or 100 nM ICI. Upon completion of the incubation
period, cells were washed with DPBS, detached (0.25% trypsin in 0.2
mM EDTA), resuspended in DPBS, counted and assessed for viability
using the trypan blue assay. Each experiment was performed at least
three times in triplicate.
Immunofluorescence studies
Cells exposed to different experimental conditions
were grown on a sterile coverglass and then fixed (absolute
methanol at −20°C), rinsed with DPBS and blocked for 30 min with 2%
BSA in DPBS. Subsequently, the samples were incubated with the
primary antibodies for 1 h at room temperature. After extensive
washes in DPBS containing 2% BSA, the samples were incubated with
the appropriate secondary antibody for 1 h and washed. Nuclei were
counterstained with 7-aminoactinomycin D (BD Pharmingen, San Diego,
CA, USA). After a final washing, the samples were mounted with
Biomeda Gel/Mount (Foster City, CA, USA) and inspected with a Zeiss
Axiophot epifluorescence microscope fitted with a color CCD camera
(Kappa GmbH, Goettingen, Germany). In each experiment, the images
were obtained under fixed settings of illumination, exposure time
and camera gain.
Cell cycle analysis
Approximately 8×105 MCF7 cells/well were
seeded in 6-well plates and treated as described for the respective
experiments. Analysis was performed using the FITC BrdU Flow kit
(BD Pharmingen) following the manufacturer’s instructions. After
the different stimuli, cells were incubated for 240 min with 10 μM
BrdU and detached using trypsin-EDTA before fixation in BD
Cytofix/Cytoperm buffer. After DNase treatment to obtain a better
exposure of the epitopes, the incorporated BrdU was detected with
the FIT-conjugated anti-BrdU-antibody. DNA was counterstained with
7-aminoactinomycin D for 30 min, then the samples were finally
resuspended in staining buffer and analyzed within 1 h. Before any
treatment, the MCF7 cells were synchronized by 24 h serum
deprivation, and then incubated for 24–48 h with vehicle, 2 nM
E2 or different concentrations of 27OHC in the absence
or presence of 100 nM ICI. Flow analysis was performed with a
BD-FACSCalibur cytometer.
Statistical analyses
The student’s t-test was used to evaluate
differences between the samples and their respective controls.
P<0.05 was considered significant. Data were analyzed using
Statistica version 6 for Windows (Statsoft Inc., USA).
Results
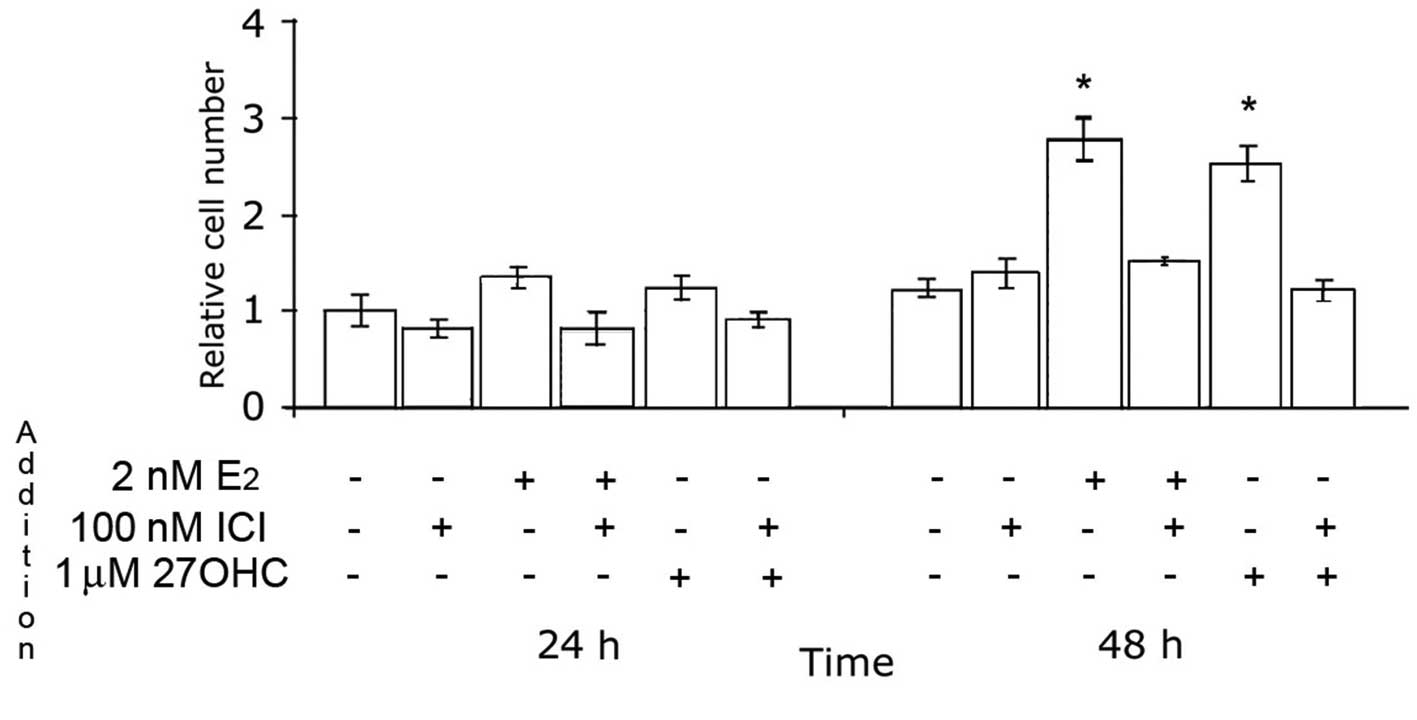
The effects of 27OHC on the proliferation of MCF7
cells in media containing low levels of serum were first analyzed.
The results are depicted in Fig.
1, and indicate that 27OHC stimulated mammary tumor cell
proliferation. The maximal stimulatory effect was obtained with 1–2
μM 27OHC, and was in the same order of magnitude as that obtained
with 2 nM E2. Similar effects of E2 and
27OHC, albeit with slower proliferation rates, were observed in
ER-positive ZR75 cells (data not shown).
In contrast to MCF7 and ZR75 cells, when MCF10 cells
were incubated under the same conditions, neither estradiol nor
27OHC had an effect on cell proliferation during the first 24 h.
Later during the exposure, 2 nM E2 still exhibited no
effects, while 1–10 μM 27OHC induced a reduction in the number of
MCF10 cells in comparison to non-treated cells (Fig. 2).
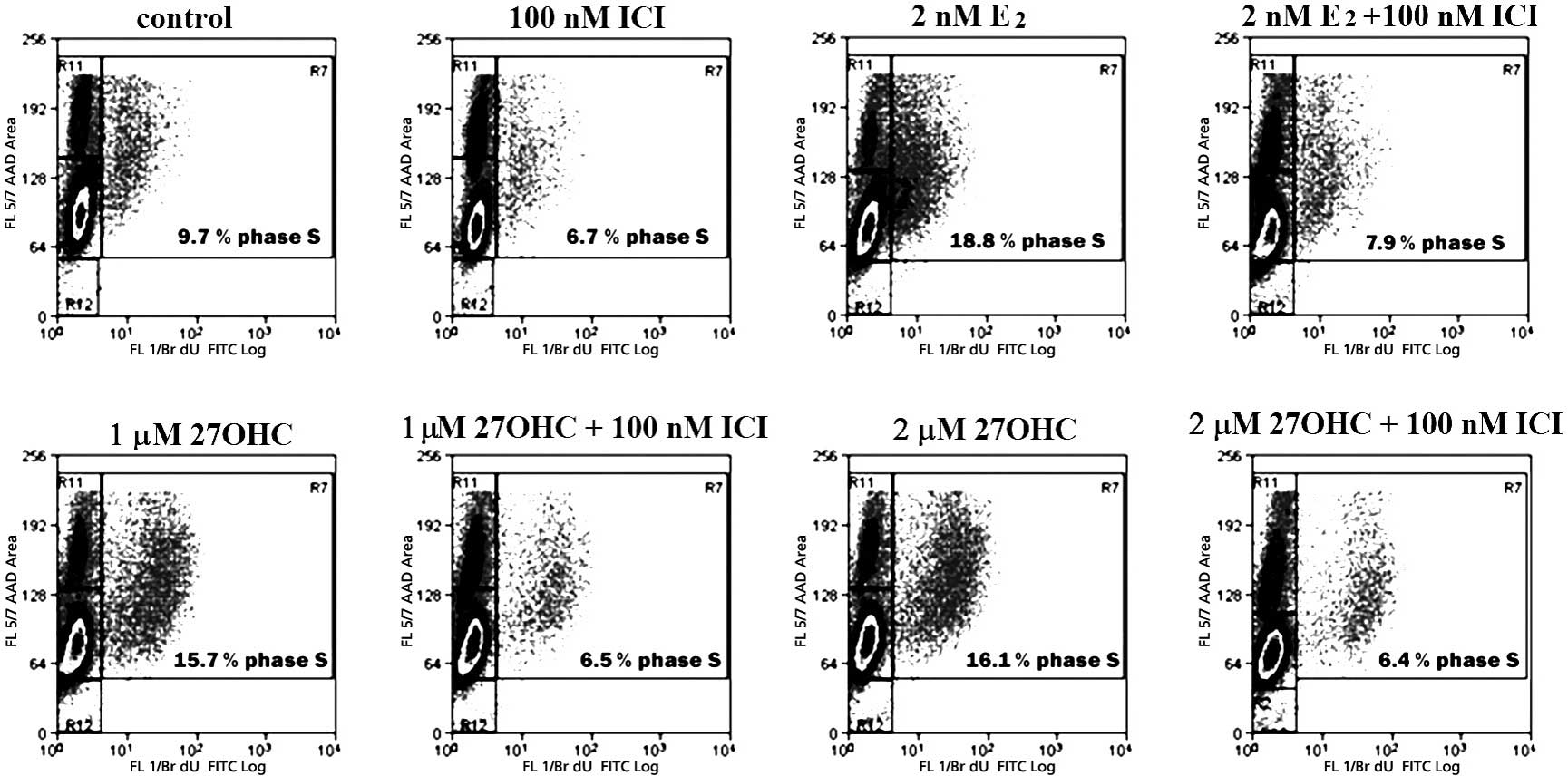
The effects of 27OHC and estradiol on the
proliferation of MCF7 cells, compared using BrdU pulses to stain
proliferating cells in the S-phase of the cell cycle, were
completely abolished by 100 nM ICI, as shown in Fig. 3.
Fig. 4 depicts a
representative cycle analysis of cells stimulated for 48 h with 2
nM E2 or with 1–2 μM 27OHC, and the respective effects
of 100 nM ICI. Stimulation with E2 or 27OHC brought a
similar number of cells into the S-phase. These effects were
completely abolished by ICI. The analyses also indicated that the
extent of apoptosis was not significantly altered within the tested
exposure duration. After 24 h of stimulus, E2 and the
novel SERM significantly increased the percentage of cells in the
S-phase. In this case, the effects were completely abolished by 100
nM ICI (data not shown).
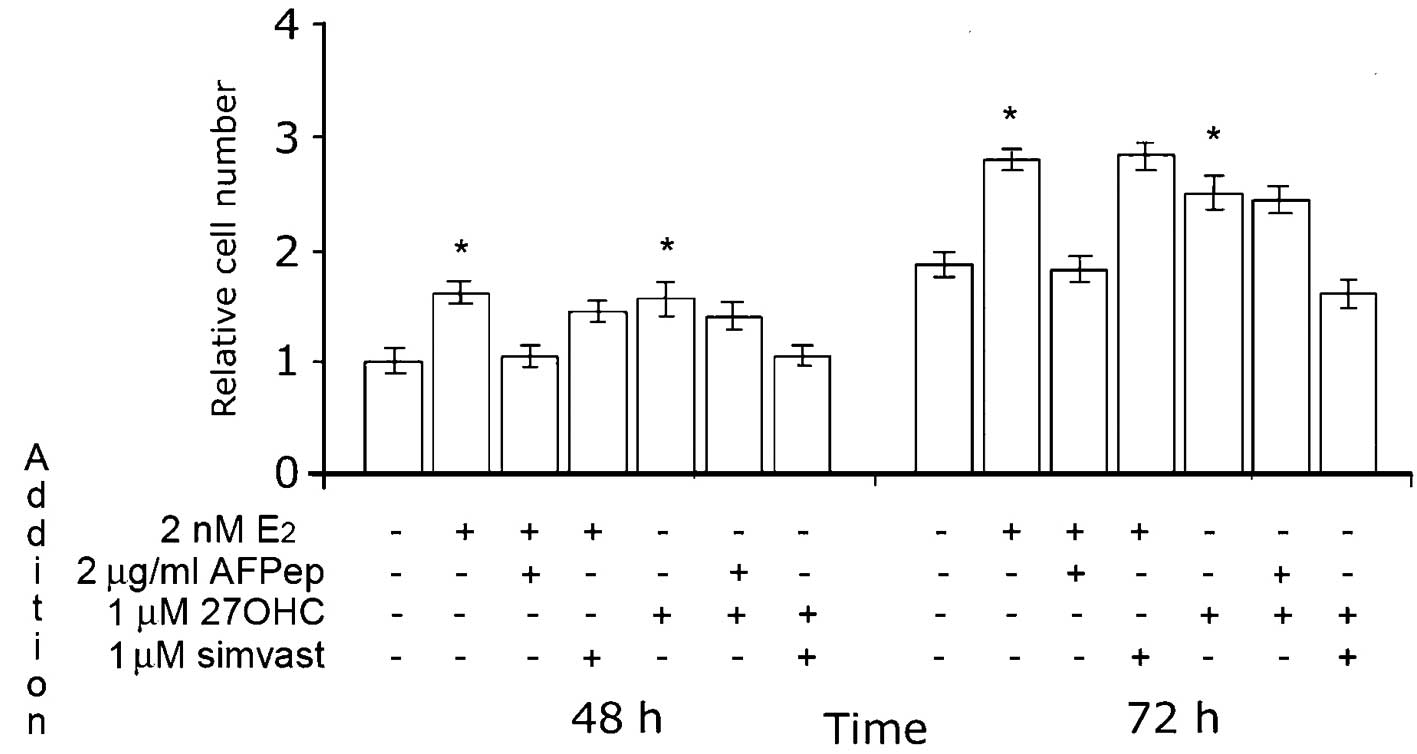
As depicted in Fig.
5, the effect of 27OHC was completely abolished by simvastatin.
The maximal effect of this HMGR inhibitor was obtained at 1 μM;
notably, the proliferation of MCF7 cells under estradiol
stimulation was not affected by the addition of simvastatin at this
concentration. Also of note, 2 μg/ml AFPep inhibited the
proliferation of MCF7 cells stimulated with 2 nM E2, but
did not affect the influence of 27OHC on cell growth.
As compared to control non-stimulated MCF7 cells,
EGFR2 immunoreactivity increased after a 15-min stimulation with 2
nM E2. This effect was abolished by 2 μg/ml AFPep. No
effects on EGFR2 reactivity were observed upon exposure to 1 μM
27OHC, independent of the presence of AFPep or simvastatin.
Fig. 6 depicts representative
fluorescence images of these experiments.
Unlike 27OHC, cholesterol added at concentrations of
up to 20 μM did not show any stimulatory effect on MCF7 cell
proliferation, as summarized in Table
I.
 | Table I.Comparative effects of 27OHC and
cholesterol on the proliferation of MCF7 cells. |
Table I.
Comparative effects of 27OHC and
cholesterol on the proliferation of MCF7 cells.
| Treatment | Proliferation
|
|---|
| 48 h | 72 h |
|---|
| Control | 1.00±0.06 | 1.51±0.05 |
| 1 μM 27OHC | 1.78±0.07a | 2.63±0.07a |
| 1 μM 27OHC + 1 μM
simvastatin | 1.04±0.08 | 1.33±0.09 |
| 5 μM cholesterol | 1.03±0.05 | 1.56±0.09 |
| 5 μM cholesterol + 1
μM simvastatin | 0.96±0.05 | 1.36±0.09 |
| 20 μM
cholesterol | 1.10±0.03 | 1.61±0.02 |
| 20 μM cholesterol + 1
μM simvastatin | 1.06±0.12 | 1.66±0.09 |
Discussion
Cholesterol is abundantly distributed in eukaryotes.
In higher animals, the main sources of circulating cholesterol are
the uptake from fat rich foods and endogenous synthesis (12). The blood of normal human subjects
contains several oxidized metabolites of cholesterol (oxysterols).
Of these, 27OHC is the most abundant species, reflecting
cholesterol saturation in the body and predicting to some extent
the responsiveness to dietary cholesterol (13). Circulating 27OHC levels increase in
cardiovascular disease (14), and
this metabolite is easily detected in macrophages isolated from
atherosclerotic lesions (15). Due
to their higher polarity and limited stuffing in cell membranes,
oxysterols share the ability to translocate faster than cholesterol
across membranes, allowing their participation in cell signaling,
lipid metabolism and vesicle transport (16). 27OHC affects some cellular
functions in macrophages (17); in
fact, the oxysterol exhibits a concentration-dependent regulation
in human macrophages: low concentrations of 27OHC, favor cell
survival, while high concentrations induce apoptosis (18). Some authors have reported that, in
macrophages and in certain intestinal cancer human cell lines,
27OHC activates the liver orphan receptor α (LXRα), inducing the
expression of efflux transporters ABCA1 and ABCG1 and thus
promoting the detoxification of cholesterol metabolites (19). Many other cells are affected by
27OHC, including vascular smooth muscle cells (20) and endothelial cells (21). Certain effects of 27OHC are
probably a consequence of its potent HMGR inhibitory activity
(22).
In the present study, we compared the effects of
27OHC and estradiol on the proliferation of mammary tumor cells in
culture. The results confirmed earlier evidence regarding a
mitogenic activity of 27OHC in estrogen-responsive breast tumor
cells. Similarly to estradiol, 1 μM 27OHC was found to stimulate
the proliferation of MCF7 cells, promoting significant changes in
the fraction of cells entering the DNA-synthesis phase in the
cell-cycle. The effects of 27OHC and estradiol on MCF7 cells were
counteracted to the same extent by the pure estrogen-antagonist ICI
182,780. Neither estradiol nor 27OHC or ICI affected the
proliferation of non-tumorigenic MCF10 cells. The stimulatory
effect of 27OHC differed from that from E2 in relation
to the sensitivity to 1 μM simvastatin and to AFPep, respectively.
In the first case, obtained at the optimal concentrations of 27OHC
and simvastatin, the results suggest that these HMGR inhibitors may
act additively, most probably causing a decrease in the
isoprenylation capacity. At the same concentration of simvastatin,
no major changes were observed in estradiol-induced MCF7
proliferation, probably because isoprenylation was not dramatically
affected (Sierralta et al, unpublished data).
In ER-positive human and canine mammary tumor cells,
AFPep is an effective inhibitor of estradiol-stimulated
proliferation (23,24). This cyclized nonapeptide disturbs
membrane receptor-tyrosine kinase signaling pathways regulated by
growth factors (25,26). By indirect immunofluorescence, we
detected an increase in EGFR2 reactivity after 2 nM estradiol. This
was abolished by AFPep; however, upon exposure to 1 μM 27OHC, no
changes in EGFR2 immunoreactivity were detected, suggesting that
estradiol and 27OHC have different mechanisms of action.
27OHC is a low-affinity ER ligand, and there remain
some doubts regarding its actual physiological relevance. As
pointed out by DuSell and McDonnell (27), during estrogen-depletion, 27OHC
affects ER by acting as a type of intracrine/paracrine modulator,
and not as a classical endocrine agent. In mammary cancer cells,
27OHC works as a partial ER agonist that recruits ERα to target
gene promoters and controls the transcription of ER target genes
(11). The ability of 27OHC to
stimulate, at physiological concentrations, the proliferation of
ERα-positive mammary-cancer cells would appear to be of major
physiological importance.
Occasionally, resistance develops during the
treatment of breast cancer with anti-estrogens and aromatase
inhibitors. Many therapy-resistant breast cancers do maintain ER
expression, indicating the continued operation of estrogen
signaling pathways. A permanently activated ER may well be the
consequence of an association with endogenously produced SERMs,
such as 27OHC. Through the metabolism of profusely available plasma
membrane cholesterol, many cells produce and secrete 27OHC in their
neighborhood, as demonstrated in the case of fibroblasts (28). The infiltration of macrophages is
an indicator of poor prognosis in patients with a mammary gland
affected by a tumor (29). It has
been demonstrated that macrophages actively increase the local
formation of 27OHC (30). Taken
together, these findings suggest that, in tissues infiltrated by
cancer, a constant stimulation of tumor cells may be caused by the
activity of local macrophages. It was previously thought that the
increased risk of breast cancer caused by obesity was mainly due to
enhanced aromatase capacity in adipose tissue, which generates
higher local concentrations of estradiol. Since obesity is
associated with hypercholesterolemia and increased 27OHC
production, a proliferative effect of this oxysterol on ER-positive
cancer cells is likely to be more intense in obese patients.
Besides binding to ER, 27OHC is considered a
potential ligand of orphan receptor LXR (31). This interaction should be taken
into consideration, since Vedin et al (32) have reported the growth-inhibitory
effects of LXR agonist GW3965 in ER-positive breast cancer cell
lines. This anti-proliferative effect was absent in ER-negative
cell lines, suggesting that ER plays an important mediator role in
the process (32).
A complete understanding of whether the final cell
response depends on the proportions of ER and LXR and the ratios of
SERM and SLXRM present at a given time in a particular tissue
requires further study. In the meantime, there is agreement that an
ample characterization of all the mechanisms involved in
estrogen-stimulated breast cancer growth is required to improve
diagnosis and the ability to predict the response of a tumor to a
specified therapy (33). As would
be expected due to the complexity of any biological system, it is
safe to predict that many more as yet unknown interacting factors
are involved in the modulation of cancer cell growth.
Acknowledgements
We are grateful to Professors H.I.
Jacobson, J. Bennett and T.T. Andersen (Albany Medical College, NY,
USA) for providing the AFPep. This study was supported by Fondecyt
Chile, Grant 1090057.
References
|
1.
|
Russo IH and Russo J: Role of hormones in
mammary cancer initiation and progression. J Mammary Gland Biol
Neoplasia. 3:49–61. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Conzen SD: Nuclear receptors and breast
cancer. Mol Endocrinol. 22:2215–2228. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Fox EM, Andrade J and Shupnik MA: Novel
actions of estrogen to promote proliferation integration of
cytoplasmic and nuclear pathways. Steroids. 74:622–627. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Madak-Erdogan Z, Kieser KJ, Kim SH, Komm
B, Katzenellenbogen JA and Katzenellenbogen BS: Nuclear and
extranuclear pathway inputs in the regulation of global gene
expression by estrogen receptors. Mol Endocrinol. 22:2116–2127.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
He J, Cheng Q and Xie W: Minireview:
nuclear receptor-controlled steroid hormone synthesis and
metabolism. Mol Endocrinol. 24:11–21. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Sasano H, Suzuki T, Nakata T and Moriya T:
New development in intracrinology of breast carcinoma. Breast
Cancer. 13:129–136. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Arpino G, Weichmann L, Osborne CK and
Schiff R: Crosstalk between the estrogen receptor and the HER
tyrosine kinase receptor family: molecular mechanism and clinical
implications for endocrine therapy resistance. Endocr Rev.
29:217–233. 2008. View Article : Google Scholar
|
|
8.
|
Hall JM and McDonnell DP: Coregulators in
nuclear estrogen receptor action: from concept to therapeutic
targeting. Mol Interv. 5:343–357. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Shelly W, Draper MW, Krishnan V, Wong M
and Jaffe RB: Selective estrogen receptor modulators: an update on
recent clinical findings. Obstet Gynecol Surv. 63:163–181.
2008.PubMed/NCBI
|
|
10.
|
Umetani M, Domoto H, Gormley AK, Yuhanna
IS, Cummins CL, Javitt NB, Korach KS, Shaul PW and Mangelsdorf DJ:
27-Hydroxycholesterol is an endogenous SERM that inhibits the
cardiovascular effects of estrogens. Nature Med. 13:1185–1192.
2007. View
Article : Google Scholar : PubMed/NCBI
|
|
11.
|
DuSell CD, Umetani M, Shaul PW,
Mangelsdorf DJ and McDonnell DP: 27-Hydroxycholesterol is an
endogenous selective estrogen receptor modulator. Mol Endocrinol.
22:65–77. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Brown AJ and Jessup W: Oxysterols and
atherosclerosis. Atherosclerosis. 142:1–28. 1999. View Article : Google Scholar
|
|
13.
|
Hirayama T, Mizokami Y, Honda A, Homma Y,
Ikegami T, Saito Y, Miyazaki T and Matsuzaki Y: Serum concentration
of 27-hydroxycholesterol predicts the effects of high-cholesterol
diet on plasma LDL cholesterol level. Hepatol Res. 39:149–156.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Babiker A, Dzeletovic S, Wiklund B,
Pettersson N, Salonen J, Nyyssönen K, Eriksson M, Diczfalusy U and
Björkhem I: Patients with atherosclerosis may have increased
circulating levels of 27-hydroxycholesterol and cholestenoic acid.
Scand J Clin Lab Invest. 65:365–375. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Hultén LM, Lindmark H, Diczfalusy U,
Björkhem I, Ottosson M, Liu Y, Bondjers G and Wiklund O: Oxysterols
present in atherosclerotic tissue decrease the expression of
lipoprotein lipase messenger RNA in human monocyte-derived
macrophages. J Clin Invest. 97:461–468. 1996.PubMed/NCBI
|
|
16.
|
Olkkonen VM and Hynynen R: Interactions of
oxysterols with membranes and proteins. Mol Aspects Med.
30:123–133. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Lemaire-Ewing S, Prunet C, Montange T,
Vejux A, Berthier A, Bessede G, Corcos L, Gambert P, Neel D and
Lizard G: Comparison of the cytotoxic, pro-oxidant and
pro-inflammatory characteristics of different oxysterols. Cell Biol
Toxicol. 21:97–114. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Riendeau V and Garenc C: Effect of
27-hydroxycholesterol on survival and death of human macrophages
and vascular smooth muscle cells. Free Radic Res. 43:1019–1028.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Li T, Chen W and Chiang JY: PXR induces
CYP27A1 and regulates cholesterol metabolism in the intestine. J
Lipid Res. 48:373–384. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Oyama T, Miyashita Y, Kinoshita K,
Watanabe H, Shirai K and Yagima T: Effect of deposited lipids in
atheromatous lesions on the migration of vascular smooth muscle
cells. J Atheroscler Thromb. 9:109–113. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Kummerow FA, Mahfouz MM, Zhou Q and Cook
LS: 27-Hydroxycholesterol causes remodeling in endothelial cell
membrane lipid composition comparable to remodeling in the failed
vein grafts of CABG patients. Life Sci. 78:958–963. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Lange Y, Ory DS, Ye J, Lanier MH, Hsu FF
and Steck TL: Effectors of rapid homeostatic responses of
endoplasmic reticulum cholesterol and
3-hydroxy-3-methylglutaryl-CoA reductase. J Biol Chem.
283:1445–1455. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Sierralta WD, Epuñán MJ, Reyes JM,
Valladares LE, Andersen TT, Bennett JA, Jacobson HI and Pino AM: A
peptide derived from α-fetoprotein inhibits the proliferation
induced by estradiol in mammary tumor cells in culture. Oncol Rep.
19:229–235. 2008.
|
|
24.
|
Sierralta WD, Epuñán MJ, Reyes JM,
Valladares LE and Pino AM: A cyclic nonapeptide derived from
alpha-fetoprotein inhibits the estradiol-induced proliferation of
mammary tumor cells in culture through the modulation of p21. Adv
Exp Med Biol. 617:463–468. 2008. View Article : Google Scholar
|
|
25.
|
Torres C, Antileo E, Epuñán MJ, Pino AM,
Valladares LE and Sierralta WD: A cyclic peptide derived from
α-fetoprotein inhibits the proliferative effects of the epidermal
growth factor and estradiol in MCF7 cells. Oncol Rep. 19:1597–1603.
2008.
|
|
26.
|
Torres CG, Pino AM and Sierralta WD: A
cyclized peptide derived from α-fetoprotein inhibits the
proliferation of ER-positive canine mammary cancer cells. Oncol
Rep. 21:1397–1404. 2009.
|
|
27.
|
DuSell CD and McDonnell DP:
27-Hydroxycholesterol: a potential endogenous regulator of estrogen
receptor signaling. Trends Pharmacol Sci. 29:510–514. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Lange Y, Steck TL, Ye J, Lanier MH, Molugu
V and Ory D: Regulation of fibroblast mitochondrial
27-hydroxycholesterol production by active plasma membrane
cholesterol. J Lipid Res. 50:1881–1888. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Steele RJ, Eremin O, Brown M and Hawkins
RA: A high macrophage content in human breast cancer is not
associated with favourable prognostic factors. Br J Surg.
71:456–458. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Javitt NB: 25R,26-Hydroxycholesterol
revisited: synthesis, metabolism and biologic roles. J Lipid Res.
43:665–670. 2002.PubMed/NCBI
|
|
31.
|
Chen W, Chen G, Head DL, Mangelsdorf DJ
and Russell DW: Enzymatic reduction of oxysterols impairs LXR
signaling in cultured cells and the livers of mice. Cell Metab.
5:73–79. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Vedin LL, Lewandowski SA, Parini P,
Gustafsson JÅ and Steffensen KR: The oxysterol receptor LXR
inhibits proliferation of human breast cancer cells.
Carcinogenesis. 30:575–579. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Culhane AC and Howlin J: Molecular
profiling of breast cancer: transcriptomic studies and beyond. Cell
Mol Life Sci. 64:3185–3200. 2007.PubMed/NCBI
|