Introduction
Cell cycle progression and cell proliferation are
controlled in part by a balance between cyclins, cyclin-dependent
kinases (Cdks) and phosphatases. Various types of human cancers are
characterized by uncontrolled cell growth which leads to poor
patient prognosis. One of the mechanisms involved in uncontrolled
cell growth is thought to be the change in basic cell cycle
regulation caused by cancer-associated mutations, overexpression of
cell cycle-regulated protein, as well as by the loss of Cdk
inhibitor expression. Cdc25 is a dual-specificity protein tyrosine
phosphatase which catalyzes the dephosphorylation and activation of
cyclin-Cdk complexes through the removal of inhibitory phosphates.
The Cdc25 phosphatase family comprises three related gene products,
namely Cdc25A, Cdc25B and Cdc25C. Cdc25A and Cdc25B cooperate with
activated Ras to induce oncogenic focus formation in rat embryonic
fibroblasts. Moreover, overexpression of Cdc25 phosphatases in
human cancers correlates with disease progression and is an
indicator of poor patient prognosis (1,2).
Taken together, these results suggest that Cdc25 phosphatases have
potential as a potent small molecular target for cancer
therapy.
In prostate cancer, Cdc25A, Cdc25B and Cdc25C are
upregulated in cancerous lesions relative to non-cancerous lesions,
and levels are increased in higher Gleason grade tumors (3–5). A
number of novel Cdc25 inhibitors from the compounds library at the
University of Pittsburgh have been screened with a focus on
compound structures, such as quinine or naphthoquinone structures
(6–8). In the present study, a more selective
naphthoquinone Cdc25 inhibitor,
7-chloro-6-(2-morpholin-4-ylethylamino) quinoline-5, 8-dione (DA
3003-2), was generated from the drug library at the University of
Pittsburgh, and its molecular mechanisms of action were
investigated using the PC-3 human prostate cancer cell line.
Materials and methods
Cell culture, chemicals and
antibodies
PC-3 human prostate cancer cells were obtained from
the American Type Culture Collection (Bethesda, MD). The cells were
cultured at 37°C in RPMI-1640 medium supplemented with 10% fetal
bovine serum in a humidified atmosphere of 5% CO2. DA
3003-2 and (2-mercaptoethanol)-3-methyl-1, 4-naphthoquinone (NSC
672121) were generated as described previously (7). These compounds were solubilized so
that the final concentration of dimethyl sulfoxide (DMSO) was
<0.1% when added to the cells. The following antibodies were
used: anti-Cdc25A (Ab3) purchased from NeoMarkers, Inc. (Fremont,
CA); anti-Cdc25B from Transduction Laboratories (Lexington, KY);
and anti-Cdc25C (C-2), anti-Cdk2 (D-12), anti-Cdc2 (17),
anti-phospho-Cdc2/Cdk2 (Tyr15), anti-cyclin A (H-432),
anti-cyclin B1 (GNS1) and anti-actin (C-2) from Santa
Cruz Biotechnology (Santa Cruz, CA).
MTT assay
The sensitivity of the cells to NSC 672121 and DA
3003-2 was determined by a microtiter assay. Cells
(4×103) were plated in 96-well microtiter plates,
cultured for 24 h and exposed continuously to 0.3–30 μM of NSC
672121 or DA 3003-2 for 48 h. The viability of the cells was
assayed by determining the color development resulting from the
reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium
bromide (MTT) spectrophotometrically at 540 nm, as described
previously (7).
Flow cytometry
PC-3 cells were plated at 5×105
cells/dish and maintained for 24 h. Cells were treated with DMSO,
NSC 672121 or DA 3003-2. After 24 h, cells were trypsinized and
washed with phosphate-buffered saline (PBS). The harvested cells
were stained with a solution containing 50 μg/ml propidium iodide
(PI) and 250 μg/ml RNase A. Flow cytometric analysis was conducted
using EPICS XL™ and XL-MCL™ systems (Beckman Coulter, Inc.).
Western blotting and
immunoprecipitation
Vehicle- or compound-treated cells were harvested by
scraping and were resuspended in lysis buffer (30 mM HEPES, 1%
Triton X-100, 10 mM NaCl, 10% glycerol, 5 mM MgCl2, 25
mM NaF, 1 mM EDTA and 0.2 mM Na3VO4, pH 7.6)
with protease inhibitors [10 μg/ml leupeptin, 10 μg/ml aprotinin
and 100 μg/ml 4-(2-aminoethyl)benzenesulfonyl fluoride]. The
samples were briefly vortexed and centrifuged at 13,000 x g for 30
min. The total protein concentration was determined using the
Bradford protein assay (BioRad, CA). To immunoprecipitate cyclin
B1, cyclin A, Cdc2 and Cdk2 proteins, 2 μg of
anti-cyclin B1, anti-cyclin A, anti-Cdc2 or anti-Cdk2
antibodies and Protein G Sepharose 4B were incubated with 1 mg of
lysates for 16 h. Beads were washed three times by vortexing with
lysis buffer at each step. Equal amounts of protein or supernatant
were resolved by SDS-PAGE and transferred to a nitrocellulose
membrane. A chemiluminescence detection system (Western Lightning™;
Perkin Elmer Life Sciences, Boston, MA) was used for immunocomplex
detection.
Results
Cdc25 inhibitor induced G2/M accumulation
in asynchronous PC-3 cells
To determine the cytotoxicity of DA 3003-2 in PC-3
cells as compared with the cytotoxic effects of the non-specific
Cdc25 inhibitor NSC 672121 [2-(2-mercaptoethanol)-3-methyl-1,
4-naphthoquinone], the MTT assay was performed. The IC50
of DA 3003-2 in PC-3 cells was 2-fold higher than that of NSC
672121, and the IC50 values of NSC 672121 and DA 3003-2
were ∼10 and 5 μM, respectively (Fig.
1). Tamura et al reported that NSC 672121 induced dual
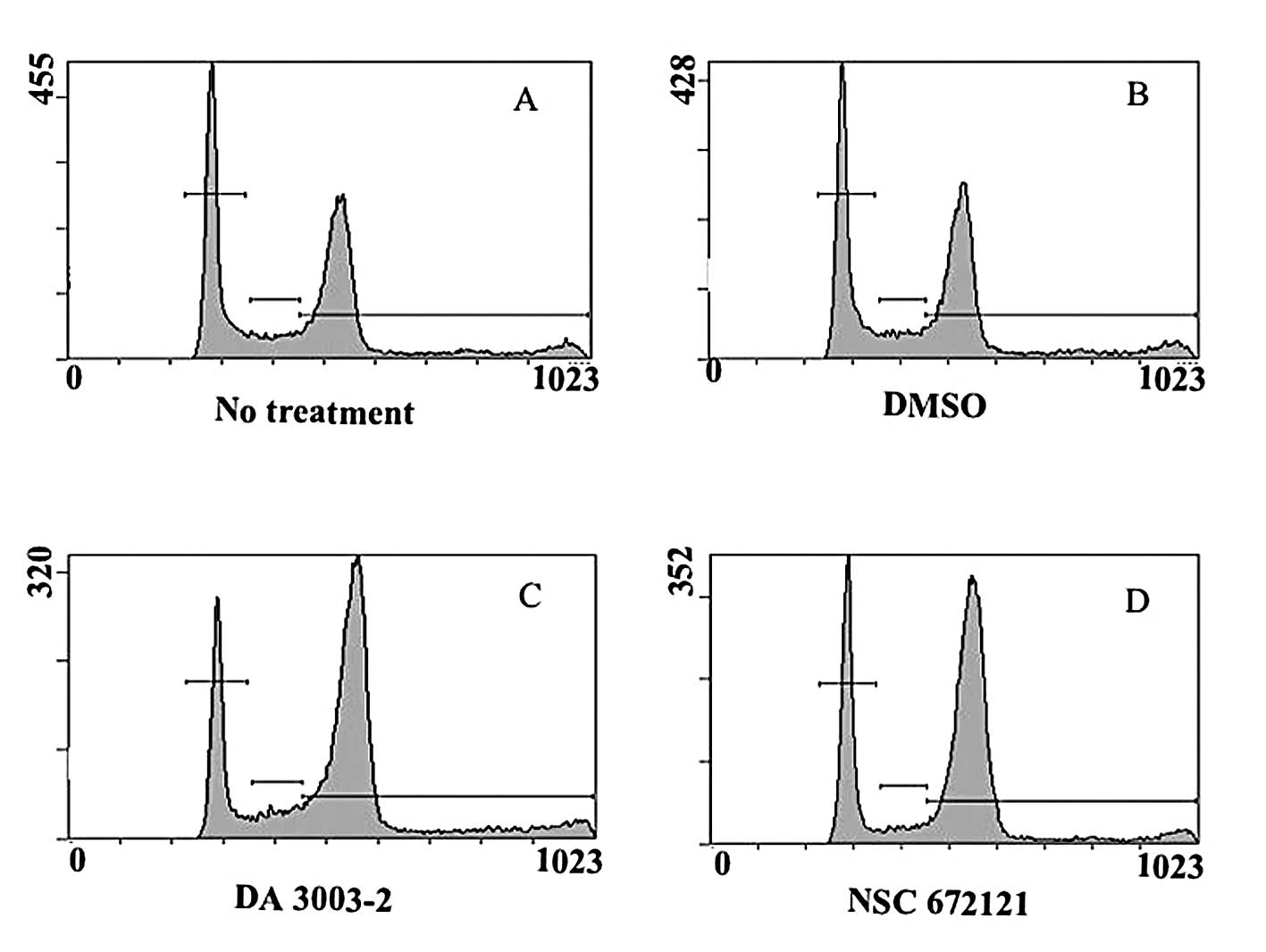
G1 and G2/M arrest in synchronized tsFT210 cells (9). Next, the changes in cell cycle
distribution induced in asynchronous PC-3 cells by NSC 672121 and
DA 3003-2 were analyzed. After 24 h, PC-3 cells had accumulated in
the G2/M phase in a concentration-dependent manner as a result of
the administration of both compounds (Fig. 2, Table
I). DA 3003-2 induced G2/M accumulation at a concentration that
was lower than that for NSC 672121 supporting the results of the
MTT assay.
 | Table I.Cell cycle distribution 24 h after
treatment with and without Cdc25 inhibitors. |
Table I.
Cell cycle distribution 24 h after
treatment with and without Cdc25 inhibitors.
| G1 | S | G2/M |
|---|
| No treatment | 35.8±2.4 | 8.2±0.4 | 52.5±2.8 |
| DMSO | 35.7±1.5 | 8.0±1.1 | 53.2±1.0 |
| DA 3003-2 | | | |
| 5 μM | 33.9±2.3 | 12.3±1.5 | 46.8±1.6 |
| 10 μM | 19.6±1.8 | 7.3±0.6 | 70.3±1.4 |
| NSC 672121 | | | |
| 10 μM | 36.3±1.0 | 7.9±0.6 | 54.9±1.8 |
| 20 μM | 25.7±2.0 | 5.5±0.7 | 67.2±1.3 |
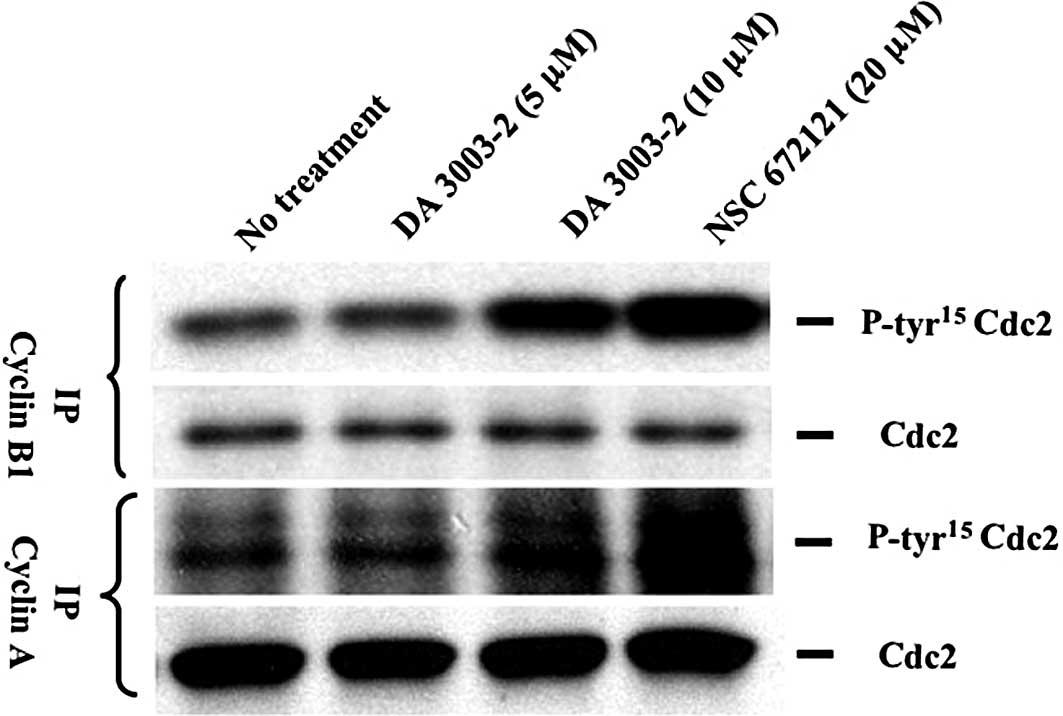
Hyperphosphorylation of Cdc2
Tyr15 in the cyclin-Cdk complex by DA 3003-2
Dephosphorylation of Cdk tyrosine by Cdc25s is
necessary for the full activation of the cyclin-Cdk complex. NSC
672121 and 6-chloro-7-(2-morpholin-4-ylethylamino)quinoline-5,
8-dione (NSC 663284), which is a regioisomer of DA 3003-2, were
previously found to increase total Cdc2 Tyr15 levels in
parallel with the inhibition of Cdc2 kinase activity. In the
present study, whether DA 3003-2 and NSC 672121 affect the Tyr15
phosphorylation status of Cdc2 in cyclin-Cdc2 complexes was
investigated using a cell-based assay. Cyclin B1 or
cyclin A were immunoprecipitated from whole cell lysates harvested
1 h after exposure to 5 or 10 μM DA 3003-2 or 20 μM NSC 672121. The
lysate proteins were separated by SDS-PAGE and immunoblotted with
antibodies to both Cdc2 phospho-Tyr15 and Cdc2. As shown
in Fig. 3, 10 μM DA 3003-2 and 20
μM NSC 672121 hyperphosphorylated Cdc2 Tyr15 in both
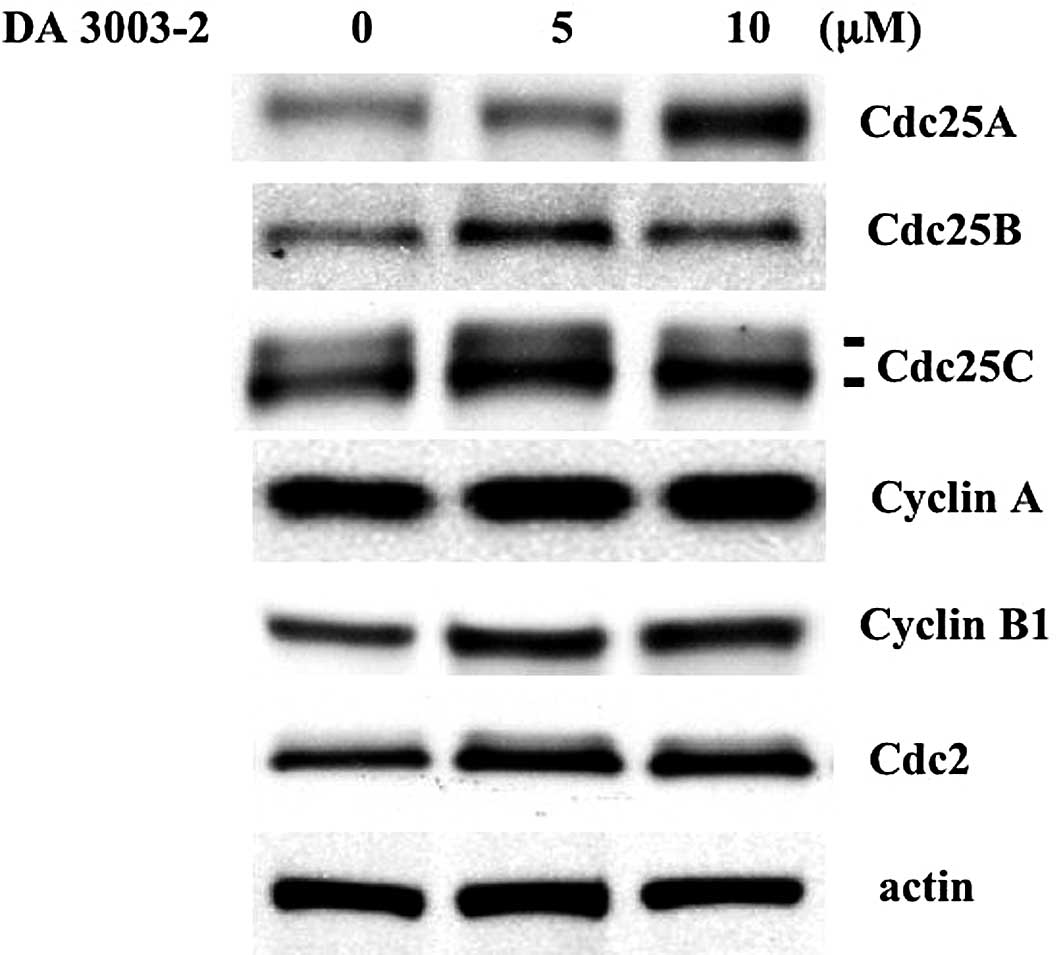
cyclin-Cdc2 complexes within 1 h. Cdc25 is an unstable protein and
is easily degraded by the proteasome pathway. Therefore, the
modulation of cell cycle regulator expression levels was
investigated using Western blotting. As shown in Fig. 4, Cdc25, G2/M cyclins and Cdc2
levels were not downregulated by DA 3003-2 administration.
Discussion
DA 3003-2 obtained from the compounds library at the
University of Pittsburgh was screened using an in vitro
phosphatase assay and was previously found to be more selective
against Cdc25B2 relative to dual phosphatase VHR or protein
tyrosine phosphatase PTP1B (6). It
has already been revealed that NSC 663284, which is a regioisomer
of DA 3003-2, inhibited Cdc25 activity by binding directly to the
Cdc25 catalytic domain (10). On
the other hand, Brisson et al suggested that the detection
of differences in the cell cycle profile of asynchronous cells
using Cdc25 inhibitors was difficult (11). In the present study, it was
revealed that G2/M accumulation was induced in asynchronous cells
using the Cdc25 inhibitor DA 3003-2 due to the hyperphosphorylation
of the G2/M cyclin-Cdk complex. A Cdc25 overexpression study will
be required to fully clarify the relationship between the
inhibition of Cdc25 activity and G2/M accumulation using a
cell-based assay. Moreover, as many different pathways feed into
cell cycle regulation, non-Cdc25-specific cellular insults are also
expected to cause G2/M arrest. However, the results of the present
study confirm the potency of the Cdc25 inhibitor.
In both early and advanced stage prostate cancer
etiology, the androgen receptor plays an important role (12,13).
Androgen ablation remains the primary course of treatment for all
patients with metastatic disease. These therapies are initially
effective. However, recurrent tumors arise within a median time of
2–3 years. The balance of androgen receptor co-regulators is
associated with the androgen refractory mechanism. Cdc25B directly
acts as the co-activator of the androgen receptor and, in contrast,
Cdc25A acts as the co-repressor (3,4).
Unfortunately, it is difficult to use previously screened Cdc25
inhibitors as molecular targeting agents for androgen co-regulators
in androgen refractory prostate cancer. This is due to the fact
that the actions of Cdc25s related to androgen receptors have
nothing to do with Cdc25 phosphatase activity. On the other hand,
several investigators have shown increased expression of cyclin
B1 in human prostate cancer (14–16).
In addition, Maddison et al demonstrated increased cyclin
B1 in poorly differentiated and androgen refractory
cancers in the TRAMP mouse model of prostate cancer (16). The levels of Cdc25C, which is an
activator of the Cdc2/cyclin B complex, increased in prostate
cancer and decreased after anti-androgen therapy. Taken together
these results suggest that G2/M transition activators, including
Cdc25, are one of the important small molecular targets for
androgen refractory prostate cancer treatment. The clinical
application of the Cdc25 inhibitor as an anti-cancer drug is
expected in the near future.
Acknowledgements
The thoughtful com ments of Professor
John S. Lazo (Department of Pharmacology, University of Pittsburgh,
Pittsburgh, PA) are sincerely appreciated. I would also like to
thank Professor Peter Wipf (Department of Chemistry, University of
Pitsburgh, Pittsburgh, PA) for providing the compounds used in the
present study.
References
|
1.
|
Kristjánsdóttir K and Rudolph J: Cdc25
phosphatases and cancer. Chem Biol. 11:1043–1045. 2004.
|
|
2.
|
Boutros R, Lobjois V and Ducommun B: CDC25
phosphatases in cancer cells: key players? Good targets? Nat Rev
Cancer. 7:495–507. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Chiu YT, Han HY, Leung SC, Yuen HF, Chau
CW, Guo Z, Qiu Y, Chan KW, Wang X, Wong YC and Ling MT: CDC25A
functions as a novel AR corepressor in prostate cancer cells. J Mol
Biol. 385:446–456. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Ngan ES, Hashimoto Y, Ma ZQ, Tsai MJ and
Tsai SY: Overexpression of Cdc25B, an androgen receptor
coactivator, in prostate cancer. Oncogene. 22:734–739. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Ozen M and Ittmann M: Increased expression
and activity of CDC25C phosphatase and an alternatively spliced
variant in prostate cancer. Clin Cancer Res. 11:4701–4706. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Lazo JS, Aslan DC, Southwick EC, Cooley
KA, Ducruet AP, Joo B, Vogt A and Wipf P: Discovery and biological
evaluation of a new family of potent inhibitors of the dual
specificity protein phosphatase Cdc25. J Med Chem. 44:4042–4049.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Lazo JS, Nemoto K, Pestell KE, Cooley K,
Southwick EC, Mitchell DA, Furey W, Gussio R, Zaharevitz DW, Joo B
and Wipf P: Identification of a potent and selective pharmacophore
for Cdc25 dual specificity phosphatase inhibitors. Mol Pharmacol.
61:720–728. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Lazo JS and Wipf P: Is Cdc25 a druggable
target? Anticancer Agents Med Chem. 8:837–842. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Tamura K, Southwick EC, Kerns J, Rosi K,
Carr BI, Wilcox C and Lazo JS: Cdc25 inhibition and cell cycle
arrest by a synthetic thioalkyl vitamin K analogue. Cancer Res.
60:1317–1325. 2000.PubMed/NCBI
|
|
10.
|
Pu L, Amoscato AA, Bier ME and Lazo JS:
Dual G1 and G2 phase inhibition by a novel, selective Cdc25
inhibitor 6-chloro-7 -[corrected]
(2-morpholin-4-ylethylamino)-quinoline-5, 8-dione. J Biol Chem.
277:46877–46885. 2002.PubMed/NCBI
|
|
11.
|
Brisson M, Nguyen T, Vogt A, Yalowich J,
Giorgianni A, Tobi D, Bahar I, Stephenson CR, Wipf P and Lazo JS:
Discovery and characterization of novel small molecule inhibitors
of human Cdc25B dual specificity phosphatase. Mol Pharmacol.
66:824–833. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Chmelar R, Buchanan G, Need EF, Tilley W
and Greenberg NM: Androgen receptor coregulators and their
involvement in the development and progression of prostate cancer.
Int J Cancer. 120:719–733. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Balk SP and Knudsen KE: AR, the cell
cycle, and prostate cancer. Nucl Recept Signal. 6:1–12.
2008.PubMed/NCBI
|
|
14.
|
Mashal RD, Lester S, Corless C, Richie JP,
Chandra R, Propert KJ and Dutta A: Expression of cell
cycle-regulated proteins in prostate cancer. Cancer Res.
56:4159–4163. 1996.PubMed/NCBI
|
|
15.
|
Kallakury BV, Sheehan CE, Ambros RA,
Fisher HA, Kaufman RP Jr, Muraca PJ and Ross JS: Correlation of
p34cdc2 cyclin-dependent kinase overexpression, CD44
downregulation, and HER-2/neu oncogene amplification with
recurrence in prostatic adenocarcinomas. J Clin Oncol.
16:1302–1309. 1998.PubMed/NCBI
|
|
16.
|
Maddison LA, Huss WJ, Barrios RM and
Greenberg NM: Differential expression of cell cycle regulatory
molecules and evidence for a ‘cyclin switch’ during progression of
prostate cancer. Prostate. 58:335–344. 2004.
|