Introduction
The largest human gene spans 2.5 Mb of the
X-chromosome and encodes the membrane cytoskeletal protein
dystrophin of 427 kDa (1).
Primary abnormalities in the dystrophin gene lead to a functional
absence of the full-length Dp427 isoform and trigger Duchenne
muscular dystrophy, a progressive neuromuscular disease of
childhood (2). The muscular
dystrophy X-linked (mdx) mouse is an established animal model of
various aspects of X-linked muscular dystrophy and is widely used
for studying fundamental mechanisms of dystrophinopathy and testing
novel therapeutic approaches to treat one of the most frequent
gender-specific diseases in humans (3). A single base substitution within
exon 23 of the dystrophin gene causes premature termination of the
dystrophin polypeptide chain in mdx mice (4). Although most individual muscles in
the mdx mouse do not represent a perfect replica of the fiber
wasting pathology observed in the highly progressive etiology of
Duchenne muscular dystrophy, certain muscle types show many
molecular and cellular alterations that are characteristic of
dystrophinopathy.
The mdx mouse shows i) a loss of the sarcolemmal
dystrophin isoform Dp427 and a drastic reduction in
dystrophin-associated glycoproteins in contractile cells (5); ii) elevated levels of serum creatine
kinase indicative of reduced muscle fiber integrity (6); iii) a varying degree of muscle
degeneration ranging from minimal effects in extraocular and
laryngeal muscle (7) to segmental
necrosis in limb muscle (8) to
severe fiber wasting in diaphragm muscle (9); iv) a high susceptibility to osmotic
shock (10) or stretch-induced
injury (11); and v) abnormal
calcium-handling (12) including
elevated cytosolic Ca2+-levels (13). These genetic, biochemical,
physiological and cell biological abnormalities have established
the mdx mouse as a suitable, albeit not precise, genocopy and
phenocopy of X-linked muscular dystrophy (3). The mdx model system has been widely
used for testing new therapeutic approaches, including myoblast
transfer therapy (14), gene
therapy (15), exon skipping
therapy (16) and pharmacological
intervention (17) and is thus a
crucial tool for the future establishment of new treatment options
(18). In contrast to
considerable phenotypic differences between young mdx muscle and
human dystrophic specimens, a large number of studies have
demonstrated that mdx muscle tissue progressively deteriorates with
age and more closely resembles the human pathology (19).
The age-related mdx pathology includes progressive
motor weakness (20), loss of
myofibers and replacement by extensive connective tissue (21–23), the presence of branched fibers
that exhibit mechanical weakening of the sarcolemma (24), a reduced life span and increased
susceptibility to spontaneous rhabdomyosarcoma (25), impaired functional and structural
recovery after injury (26), and
a decline in regenerative potential and alterations in the crucial
mTOR signaling pathway, which is of central importance for muscle
development, muscle regeneration, and muscle growth in response to
nutrients, growth factors and exercise (27). Thus, since senescent mdx muscle
tissues appear to represent a more suitable dystrophic phenotype,
it was of interest to determine global changes in the protein
complement during the natural aging process of mdx muscle tissue.
This report shows the findings of a comparative proteomic analysis
of severely affected diaphragm muscle from 8-week, 12-month and
22-month dystrophic specimens.
Materials and methods
Chemicals and materials
Materials and electrophoresis-grade chemicals for
the proteomic analysis of muscle proteins were purchased from
Amersham Biosciences/GE Healthcare (Little Chalfont, UK). For
protein digestion, sequencing grade-modified trypsin was obtained
from Promega (Madison, WI). Chemiluminescence substrate and
protease inhibitors were from Roche Diagnostics (Mannheim,
Germany). Primary antibody to collagen VI and secondary
peroxidase-conjugated antibodies were from Abcam (Cambridge, UK)
and Chemicon International (Temecula, CA), respectively. All other
chemicals used were of analytical grade and were purchased from
Sigma Chemical Co. (Dorset, UK).
Preparation of crude muscle extracts from
aged mdx mice
The mdx mouse is missing the membrane cytoskeletal
protein Dp427 due to a point mutation in the dystrophin gene
(4) and the severely affected mdx
diaphragm muscle represents an established animal model of Duchenne
muscular dystrophy (9).
Dystrophic diaphragm muscle from 8-week, 12-month and 22-month mdx
mice and normal tissues from age-matched C57 mice were obtained
from the bioresource unit of the University of Bonn (26). Mice were kept under standard
conditions and all procedures were performed in accordance with
German guidelines on the use of animals for scientific experiments.
Animals were sacrificed by cervical dislocation and muscle tissues
were quickly removed and quick-frozen in liquid nitrogen. For the
proteomic analysis of mdx tissue, specimens were shipped to Ireland
on dry ice and stored at −80°C prior to usage. In order to obtain
diaphragm protein extracts, 4 dystrophic muscle specimens from each
age group were pulverized by grinding tissue pieces in liquid
nitrogen using a mortar and pestle. Ground muscle powder was
solubilized in lysis buffer with the ratio of 100 mg wet weight to
1 ml lysis buffer [7 M urea, 2 M thiourea, 4% CHAPS, 2% IPG buffer
pH 3–10, 2% (w/v) DTT]. To prevent excess protein degradation, the
lysis buffer was supplemented with a freshly prepared protease
inhibitor cocktail (28).
Following gentle rocking for 60 min, suspensions were centrifuged
at 4°C for 20 min at 20,000 x g and the protein concentration
determined (29).
Fluorescence gel electrophoretic
analysis
For the separation of individual muscle protein
species, two-dimensional gel electrophoresis was carried out by
previously optimized methodology using first dimension isoelectric
focusing with pH 3–10 strips and second dimension slab gel
electrophoresis with 500 μg protein/ gel (28–30). Twelve slab gels were run in
parallel at 0.5 W/gel for 60 min and then 15 W/gel until the blue
dye front had disappeared from the bottom of the gel.
Post-electrophoretic staining for the total protein profile was
performed with the fluorescent dye ruthenium II tris
bathophenanthroline disulfonate (RuBPs). A stock solution of RuBPs
dye was prepared as described previously by Rabilloud et al
(31). Following fixation for 30
min in 30% ethanol and 10% acetic acid, gels were washed 3 times
for 30 min in 20% ethanol and then stained for 6 h in 20% (v/v)
ethanol containing 2 μM of ruthenium chelate. Gels were
re-equilibrated twice for 10 min in distilled water and destained
overnight in 40% ethanol and 10% acetic acid prior to imaging
(32). Fluorescently labelled
proteins were visualized using a Typhoon Trio variable mode imager
(Amersham Biosciences/ GE Healthcare). Gel analysis was performed
with Progenesis 2D analysis software (Nonlinear Dynamics, Newcastle
upon Tyne, UK) and protein spots with a significant change in
abundance were identified by mass spectrometry.
Mass spectrometric identification of
muscle-associated proteins
Protein identification was performed with 2D protein
spots from Coomassie-stained pick gels, following counter staining
of RuBPs-labelled analytical gels. Electrospray ionization LC-MS/MS
analysis was carried out as previously described in detail
(29). Previously standardized
in-gel tryptic digestion protocols were employed for the
reproducible generation of peptides for mass spectrometric analysis
on a Model 6340 Ion Trap LC/MS apparatus from Agilent Technologies
(Santa Clara, CA). Database searches were carried out using Mascot
MS/ MS Ion search. Criterion for each search was set at i) species
Mus musculus, ii) two missed cleavages by trypsin, iii) variable
modification: oxidation of methioine, iv) fixed modification:
carboxymethylation of cysteines and v) mass tolerance of precursor
ions ±2 Da and product ions ±1 Da. Verification of key proteomic
findings was carried out by comparative immunoblot analysis
(28).
Results
Gel electrophoretic analysis of aged mdx
diaphragm muscle
Fluorescence high-resolution 2D gel electrophoresis
in combination with MS analysis was used to detect potential
differences in aging-related protein expression patterns in
severely dystrophic diaphragm muscle from mdx mice. As summarized
in Fig. 1, gels representing 4
biological repeats of 8-week, 12-month and 22-month mdx diaphragm
muscle were analyzed. The overall 2D spot patterns of differently
aged dystrophic preparations were relatively comparable; requiring
therefore detailed denitometric analyses for the determination of
significant differences in individual muscle proteins. With the
help of a Typhoon Trio variable imager and Progenesis 2-D analysis
software, individual muscle proteomes separated on 12 different
gels were compared. Panels DIA MDX 1–4, DIA MDX 5–8 and DIA MDX
9–12 represent 8-week, 12-month and 22-month diaphragm muscle
preparations, respectively. The detailed proteomic survey of
dystrophic diaphragm muscle tissue identified distinct changes in a
variety of protein species.
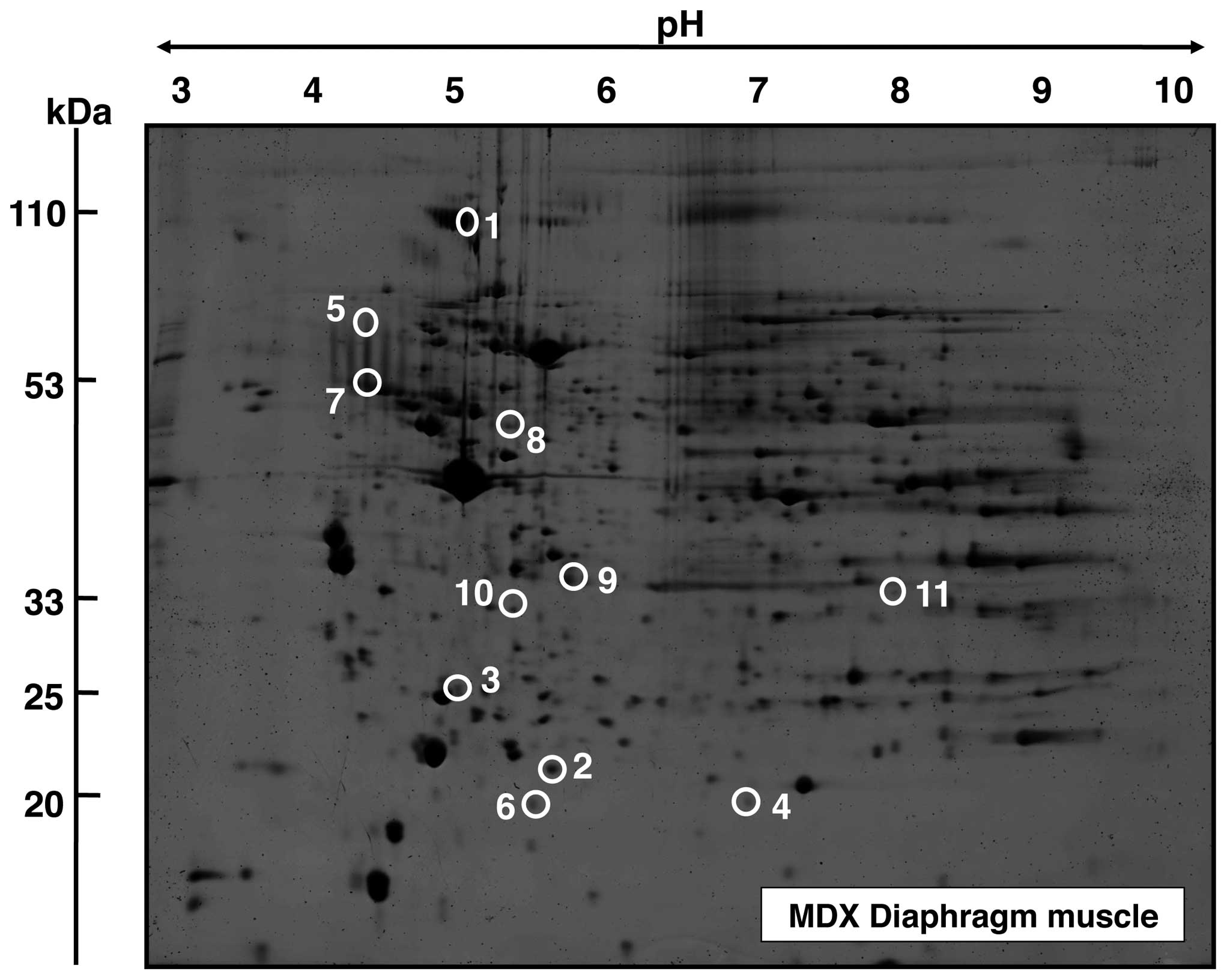
Proteomic analysis of protein alterations
in aged mdx diaphragm muscle
A representative fluorescent 2D master gel of mdx
diaphragm muscle is shown in Fig.
2. The overall number and degree of age-related changes was
striking in diaphragm muscle. Skeletal muscle proteins that
exhibited significant alterations in expression levels are marked
by circles and are numbered 1 to 11 in 2D gels of diaphragm muscle.
The mass spectrometric identification of these altered protein
species is catalogued in Table I.
Listed are the names of the identified muscle-associated proteins,
their international accession number, pI-values, their
relative molecular masses, the number of matched peptide sequences,
percentage sequence coverage, Mascot scores, and fold-change of
individual proteins affected in dystrophin-deficient tissue during
aging.
 | Table IThe identified proteins that exhibit
a drastic change in abundance during aging of the severely
dystrophic mdx diaphragm muscle. |
Table I
The identified proteins that exhibit
a drastic change in abundance during aging of the severely
dystrophic mdx diaphragm muscle.
| Spot no. | Protein name | Protein accession
no. | Isoelectric point,
pI | Molecular mass
(Da) | Peptides, n | Coverage (%) | Mascot score | Fold-change 8 w-12
m | Fold-change 8 w-22
m |
|---|
| 1 | Collagen α-1(VI)
chain | NP034063 | 5.20 | 109,582 | 12 | 15 | 259 | 3.6 | 6.3 |
| 2 | Dermatopontin | NP062733 | 4.70 | 24,559 | 4 | 21 | 122 | 5.4 | 6.1 |
| 3 | Ubiquitin
carboxyl-terminal hydrolase UCHL1 | AAD51029 | 5.33 | 25,170 | 4 | 30 | 83 | 3.5 | 4.1 |
| 4 | αB-crystallin | NP034094 | 6.76 | 20,056 | 7 | 38 | 113 | 3.7 | 4 |
| 5 | Actinin, α2 | AAK64510 | 5.34 | 104,447 | 4 | 5 | 196 | 4.3 | 3.6 |
| 6 | Ferritin heavy
chain | NP034369 | 5.53 | 21,227 | 3 | 18 | 50 | 2.6 | 2.6 |
| 7 | Vimentin | CAA39807 | 5.06 | 53,747 | 14 | 37 | 140 | 2.5 | 2.5 |
| 8 | Fibrinogen, γ
chain | NP598623 | 5.54 | 50,056 | 4 | 12 | 65 | 1.9 | 2.5 |
| 9 | Mimecan | NP032786 | 5.52 | 34,339 | 5 | 19 | 293 | 1.8 | 2.2 |
| 10 | Apolipoprotein
E | AAA37252 | 5.82 | 33,206 | 8 | 32 | 169 | 2 | 1.5 |
| 11 | Myozenin-1 | NP067483 | 8.57 | 31,438 | 9 | 54 | 174 | 0.3 | 0.3 |
Mass spectrometrically identified
proteins with an altered abundance in mdx diaphragm muscle
Protein species with a changed concentration in mdx
diaphragm muscle ranged in molecular mass from 20 kDa
(αB-crystallin) to 110 kDa (collagen) and covered a pI-range
from 4.7 (dermatopontin) to 8.6 (myozenin). As presented in
Fig. 2 and Table I, an increased concentration was
established for the α-1(VI) chain of collagen (spot 1), the
extracellular matrix protein dermatopontin (spot 2), the enzyme
ubiquitin carboxyl-terminal hydrolase (spot 3), the small heat
shock protein αB-crystallin (spot 4), α-2 actinin (spot 5),
ferritin heavy chain (spot 6), vimentin (spot 7), the γ chain of
fibrinogen (spot 8) mimecan (spot 9) and apolipoprotein E (spot
10). Spot 11 representing myozenin was shown to be decreased in
aged mdx diaphragm muscle.
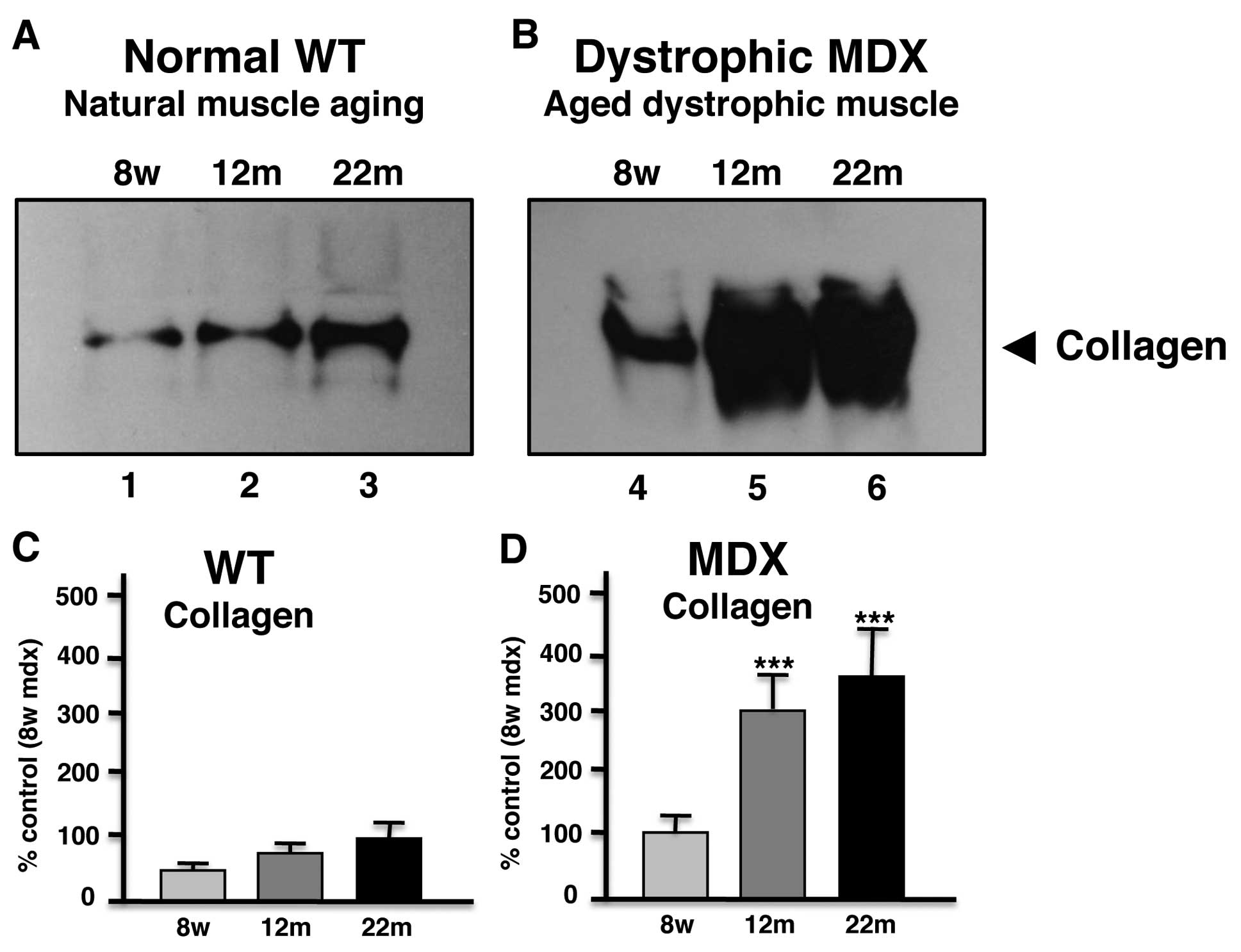
Immunoblot analysis of collagen in aged
mdx diaphragm muscle
In order to independently verify the most drastic
alteration in aged mdx diaphragm as revealed by proteomics,
comparative immunoblotting was used. As shown in Fig. 3, immunodecoration of gel
electrophoretically separated normal and mdx diaphragm of varying
age showed a general increase of collagen in 12-month and 22-month
muscle preparations. However, aged dystrophic mdx diaphragm muscle
exhibited a significantly higher increase in collagen as compared
to aged normal muscle.
Discussion
Animal models that mimic neuromuscular disorders
play a crucial role in basic and applied myology. Naturally
occurring or genetically engineered model systems are widely used
for studying fundamental aspects of molecular and cellular
pathogenesis, as well as the evaluation of novel therapeutic
approaches (33). Ideally, an
animal model of a genetic disorder should: i) exhibit similar
primary abnormalities and secondary downstream alterations as seen
in the corresponding human disease; ii) closely develop most of the
multifactorial features observed in complex human pathologies; iii)
resemble the pathogenesis of the human disease in onset,
progression and severity; iv) show sufficient similarities to human
metabolism, physiology and immune responses so that these
biological factors do not have a major differentiating influence on
disease progression in animal models vs. patients; v) is easy to
breed and house at a reasonable cost; vi) be suitable for genetic
manipulations and the facilitation of physiological and surgical
procedures; and vii) be large enough to yield sufficient amounts of
tissue specimens for extended biological analyses (34). Since an important bioethical
objection with respect to the humane and responsible use of animal
models in biomedical research is often associated with the usage of
larger animals, small rodents are the most frequently used
alternatives as genetic model systems. In the case of one the most
progressive genetic disease of the neuromuscular system, Duchenne
muscular dystrophy, the naturally occurring dystrophic mdx mouse
has been employed in a large number of studies (3,35).
In analogy to findings of previous mass
spectrometry-based proteomic studies of contractile tissues from
young or mature mdx mice (36)
which included hindlimb muscle (37–39), extraocular muscle (30), diaphragm (16,28,40) and heart (29,41), we have here carried out a
comparative proteomic survey of diaphragm muscle from 8-week,
12-month and 22-month dystrophic specimens. Dystrophic diaphragm
muscle showed altered expression levels in 11 proteins during
skeletal muscle aging. Changes in the diaphragm proteome from aged
mdx mice suggested elevated levels of fibrosis, an intensified
stress response and an increase in cytoskeletal elements possibly
compensating the lack of dystrophin. These proteomic findings agree
with the idea of more extensively perturbed protein expression
levels in dystrophin-deficient diaphragm fibers as compared to
other mdx muscle systems (36).
The approximately 6-fold increase of collagen and
dermatopontin in senescent mdx diaphragm muscle are important
proteomic findings and reflect the dystrophic status of this muscle
type. The dramatic increase of collagen in mdx diaphragm was
clearly confirmed by immunoblot analysis, verifying the findings
from the mass spectrometric investigation presented here. Although
it is clearly established that collagen levels increase in the
skeletal muscle extracellular matrix during the natural aging
process (42), the dystrophic
phenotype shows an exacerbated age-related accumulation of collagen
α-1(VI) chain. Collagen is the main protein component of connective
tissue and is especially enriched in the endomysium of skeletal
muscles. Increased collagen protein levels agree with previously
reported greater amounts of collagen mRNA in the mdx diaphragm
(43) and support the idea of
severe fibrosis in the mdx diaphragm (44,45). The non-collagenous extracellular
matrix protein dermatopontin is involved in cell-matrix
interactions and matrix assembly (46) and also named tyrosine-rich acidic
matrix protein (TRAMP) (47).
TRAMP appears to regulate interactions of TGF-β, decorin and
fibronectin (48). Its greater
abundance in mdx diaphragm is probably due to increased demands for
collagen matrix organization within dystrophic muscle tissues.
Increased levels of fibrinogen in aged mdx diaphragm, as shown here
by proteomics, agree with a previous study by Vidal et al
(49). Fibrinogen seems to play a
key role in fibrosis via a TGF-β/alternative macrophage activation
pathway in dystrophinopathy (49).
The increase of αB-crystallin and vimentin suggests
an increased cellular stress response and an upregulation of
cytoskeletal elements in dystrophic muscle, respectively, which
agrees with previous proteomic studies (28). In the future, it will be
interesting to define the potential pathobiochemical role of newly
identified biomarkers of muscular dystrophy in aged mdx diaphragm
muscle, including the deubiqutinating enzyme ubiquitin
carboxyl-terminal hydrolase, the microfilament protein actinin and
its binding protein myozenin, the intracellular iron storage
component ferritin, the connective tissue protein mimecan, and the
triglyceride transporter apolipoprotein E. Overall, the proteomic
results presented here suggest that the aged mdx diaphragm, which
exhibits severe respiratory impairment following fibrosis (50), is a highly suitable model system
for studying the molecular pathogenesis of Duchenne muscular
dystrophy. Collagen and dermatopontin should be considered as
suitable biomarker candidates for evaluating the degree of fibrosis
and tissue scaring in dystrophinopathy.
Abbreviations:
|
MS
|
mass spectrometry;
|
|
mdx
|
muscular dystrophy X-linked;
|
|
2D
|
two-dimensional;
|
|
RuBPs
|
ruthenium tris bathophenanthroline
disulfonate
|
Acknowledgements
The research was supported by project
grants from the Muscular Dystrophy Ireland and Duchenne Ireland,
and a Hume scholarship from NUI Maynooth, as well as equipment
grants from the Irish Health Research Board and the Higher
Education Authority.
References
|
1.
|
E Le RumeurSJ WinderJF HubertDystrophin:
more than just the sum of its partsBiochim Biophys
Acta180417131722201020472103
|
|
2.
|
K BushbyR FinkelDJ BirnkrantLE CasePR
ClemensL CripeA KaulK KinnettC McDonaldS PandyaDiagnosis and
management of Duchenne muscular dystrophy, part 1: diagnosis, and
pharmacological and psychosocial managementLancet
Neurol97793201010.1016/S1474-4422(09)70271-619945913
|
|
3.
|
M DurbeejKP CampbellMuscular dystrophies
involving the dystrophin-glycoprotein complex: an overview of
current mouse modelsCurr Opin Genet
Dev12349361200210.1016/S0959-437X(02)00309-X12076680
|
|
4.
|
P SicinskiY GengAS Ryder-CookEA BarnardMG
DarlisonPJ BarnardThe molecular basis of muscular dystrophy in the
mdx mouse: a point
mutationScience24415781580198910.1126/science.26624042662404
|
|
5.
|
K OhlendieckKP
CampbellDystrophin-associated proteins are greatly reduced in
skeletal muscle from mdx miceJ Cell
Biol11516851694199110.1083/jcb.115.6.16851757468
|
|
6.
|
G BulfieldWG SillerPA WightKJ MooreX
chromosome-linked muscular dystrophy (mdx) in the mouseProc Natl
Acad Sci USA8111891192198410.1073/pnas.81.4.11896583703
|
|
7.
|
MJ MarquesR FerrettiVU VomeroE MinatelHS
NetoIntrinsic laryngeal muscles are spared from myonecrosis in the
mdx mouse model of Duchenne muscular dystrophyMuscle
Nerve35349353200710.1002/mus.2069717143878
|
|
8.
|
LF TorresLW DuchenThe mutant mdx:
inherited myopathy in the mouse. Morphological studies of nerves,
muscles and
end-platesBrain110269299198710.1093/brain/110.2.2693567525
|
|
9.
|
HH StedmanHL SweeneyJB ShragerHC MaguireRA
PanettieriB PetrofM NarusawaJM LeferovichJT SladkyAM KellThe mdx
mouse diaphragm reproduces the degenerative changes of Duchenne
muscular dystrophyNature352536539199110.1038/352536a01865908
|
|
10.
|
A MenkeH JockuschDecreased osmotic
stability of dystrophin-less muscle cells from the mdx
mouseNature3496971199110.1038/349069a01985268
|
|
11.
|
GS LynchJA RafaelJS ChamberlainJA
FaulknerContraction-induced injury to single permeabilized muscle
fibers from mdx, transgenic mdx, and control miceAm J Physiol Cell
Physiol279C1290C1294200011003610
|
|
12.
|
DG AllenOL GervasioEW YeungNP
WhiteheadCalcium and the damage pathways in muscular dystrophyCan J
Physiol Pharmacol888391201020237582
|
|
13.
|
N MalloukV JacquemondB AllardElevated
subsarcolemmal Ca2+ in mdx mouse skeletal muscle fibers
detected with Ca2+-activated K+ channelsProc
Natl Acad Sci USA97495049552000
|
|
14.
|
TA PartridgeJE MorganGR CoultonEP
HoffmanLM KunkelConversion of mdx myofibres from
dystrophin-negative to -positive by injection of normal
myoblastsNature337176179198910.1038/337176a02643055
|
|
15.
|
DJ WellsKE WellsGene transfer studies in
animals: what do they really tell us about the prospects for gene
therapy in DMD?Neuromuscul
Disord12S11S22200210.1016/S0960-8966(02)00077-912206790
|
|
16.
|
P DoranSD WiltonS FletcherK
OhlendieckProteomic profiling of antisense-induced exon skipping
reveals reversal of pathobiochemical abnormalities in dystrophic
mdx
diaphragmProteomics9671685200910.1002/pmic.20080044119132684
|
|
17.
|
CF SpurneyH Gordish-DressmanAD GuerronA
SaliGS PandeyR RawatJH Van Der MeulenHJ ChaEE PistilliTA
PartridgePreclinical drug trials in the mdx mouse: assessment of
reliable and sensitive outcome measuresMuscle
Nerve39591602200910.1002/mus.2121119260102
|
|
18.
|
TA PartridgeImpending therapies for
Duchenne muscular dystrophyCurr Opin
Neurol24415422201110.1097/WCO.0b013e32834aa3f121892079
|
|
19.
|
JP LefaucheurC PastoretA SebillePhenotype
of dystrophinopathy in old mdx miceAnat
Rec2427076199510.1002/ar.10924201097604983
|
|
20.
|
GS LynchRT HinkleJS ChamberlainSV BrooksJA
FaulknerForce and power output of fast and slow skeletal muscles
from mdx mice 6–28 months oldJ Physiol535591600200111533147
|
|
21.
|
C PastoretA SebilleAge-related differences
in regeneration of dystrophic (mdx) and normal muscle in the
mouseMuscle Nerve1811471154199510.1002/mus.8801810117659109
|
|
22.
|
C PastoretA SebilleMDX mice show
progressive weakness and muscle deterioration with ageJ Neurol
Sci12997105199510.1016/0022-510X(94)00276-T7608742
|
|
23.
|
MA WineingerRT AbreschSA WalshGT
CarterEffects of aging and voluntary exercise on the function of
dystrophic muscle from mdx miceAm J Phys Med
Rehabil772027199810.1097/00002060-199801000-000049482375
|
|
24.
|
SI HeadBranched fibres in old dystrophic
mdx muscle are associated with mechanical weakening of the
sarcolemma, abnormal Ca2+ transients and a breakdown of
Ca2+ homeostasis during fatigueExp
Physiol95641656201010.1113/expphysiol.2009.05201920139167
|
|
25.
|
JS ChamberlainJ MetzgerM ReyesD TownsendJA
FaulknerDystrophin-deficient mdx mice display a reduced life span
and are susceptible to spontaneous rhabdomyosarcomaFASEB
J2121952204200710.1096/fj.06-7353com17360850
|
|
26.
|
A IrintchevM ZweyerA WernigImpaired
functional and structural recovery after muscle injury in
dystrophic mdx miceNeuromuscul
Disord7117125199710.1016/S0960-8966(96)00422-19131653
|
|
27.
|
E MouiselA VignaudC HourdeG Butler-BrowneA
FerryMuscle weakness and atrophy are associated with decreased
regenerative capacity and changes in mTOR signaling in skeletal
muscles of venerable (18–24-month-old) dystrophic mdx miceMuscle
Nerve41809818201020151467
|
|
28.
|
P DoranG MartinP DowlingH JockuschK
OhlendieckProteome analysis of the dystrophin-deficient MDX
diaphragm reveals a drastic increase in the heat shock protein
cvHSPProteomics646104621200610.1002/pmic.20060008216835851
|
|
29.
|
C LewisH JockuschK OhlendieckProteomic
profiling of the dystrophin-deficient MDX heart reveals drastically
altered levels of key metabolic and contractile proteinsJ Biomed
Biotechnol2010648501201010.1155/2010/64850120508850
|
|
30.
|
C LewisK OhlendieckProteomic profiling of
naturally protected extraocular muscles from the
dystrophin-deficient mdx mouseBiochem Biophys Res
Commun39610241029201010.1016/j.bbrc.2010.05.05220471957
|
|
31.
|
T RabilloudJM StrubS LucheA van
DorsselaerJ LunardiA comparison between Sypro Ruby and ruthenium II
tris (bathophenanthroline disulfonate) as fluorescent stains for
protein detection in
gelsProteomics1699704200110.1002/1615-9861(200104)1:5%3C699::AID-PROT699%3E3.0.CO;2-C11678039
|
|
32.
|
J GannonL StauntonK O'ConnellP DoranK
OhlendieckPhosphoproteomic analysis of aged skeletal muscleInt J
Mol Med2233422008
|
|
33.
|
M VainzofD Ayub-GuerrieriPC OnofrePC
MartinsVF LopesD ZilberztajnLS MaiaK SellLU YamamotoAnimal models
for genetic neuromuscular diseasesJ Mol
Neurosci34241248200810.1007/s12031-007-9023-918202836
|
|
34.
|
JL GuenetAnimal models of human genetic
diseases: do they need to be faithful to be useful?Mol Genet
Genomics286120201110.1007/s00438-011-0627-y21547562
|
|
35.
|
GB BanksJS ChamberlainThe value of
mammalian models for duchenne muscular dystrophy in developing
therapeutic strategiesCurr Top Dev
Biol84431453200810.1016/S0070-2153(08)00609-119186250
|
|
36.
|
C LewisS CarberryK OhlendieckProteomic
profiling of X-linked muscular dystrophyJ Muscle Res Cell
Motil30267269200910.1007/s10974-009-9197-620082121
|
|
37.
|
Y GeMP MolloyJS ChamberlainPC
AndrewsProteomic analysis of mdx skeletal muscle: Great reduction
of adenylate kinase 1 expression and enzymatic
activityProteomics318951903200310.1002/pmic.20030056114625851
|
|
38.
|
P DoranP DowlingJ LohanK McDonnellS
PoetschK OhlendieckSubproteomics analysis of
Ca2+-binding proteins demonstrates decreased
calsequestrin expression in dystrophic mouse skeletal muscleEur J
Biochem271394339522004
|
|
39.
|
D Gardan-SalmonJM DixonSM LonerganJT
SelsbyProteomic assessment of the acute phase of dystrophin
deficiency in mdx miceEur J Appl
Physiol11127632773201110.1007/s00421-011-1906-321409400
|
|
40.
|
P DoranP DowlingP DonoghueM BuffiniK
OhlendiecReduced expression of regucalcin in young and aged mdx
diaphragm indicates abnormal cytosolic calcium handling in
dystrophin-deficient muscleBiochim Biophys
Acta1764773785200610.1016/j.bbapap.2006.01.007
|
|
41.
|
MK GulstonDV RubtsovHJ AthertonK ClarkeKE
DaviesKS LilleyJL GriffinA combined metabolomic and proteomic
investigation of the effects of a failure to express dystrophin in
the mouse heartJ Proteome
Res720692077200810.1021/pr800070p18386883
|
|
42.
|
TW KragstrupM KjaerAL MackeyStructural,
biochemical, cellular, and functional changes in skeletal muscle
extracellular matrix with agingScand J Med Sci
Sports21749757201110.1111/j.1600-0838.2011.01377.x22092924
|
|
43.
|
G GoldspinkK FernandesPE WilliamsDJ
WellsAge-related changes in collagen gene expression in the muscles
of mdx dystrophic and normal miceNeuromuscul
Disord4183191199410.1016/0960-8966(94)90019-17919967
|
|
44.
|
KM GrahamR SinghG MillmanG MalnassyF
GattiK BruemmerC StefanskiH CurtisJ SestiCG CarlsonExcessive
collagen accumulation in dystrophic (mdx) respiratory musculature
is independent of enhanced activation of the NF-kappaB pathwayJ
Neurol Sci2944350201010.1016/j.jns.2010.04.00720471037
|
|
45.
|
F TrenszS HarounA CloutierMV RichterG
GrenierA muscle resident cell population promotes fibrosis in
hindlimb skeletal muscles of mdx mice through the Wnt canonical
pathwayAm J Physiol Cell
Physiol299C939C947201010.1152/ajpcell.00253.201020810909
|
|
46.
|
O OkamotoS FujiwaraDermatopontin, a novel
player in the biology of the extracellular matrixConnect Tissue
Res47177189200610.1080/0300820060084656416987749
|
|
47.
|
EG ForbesAD CronshawJR MacBeathDJ
HulmesTyrosine-rich acidic matrix protein (TRAMP) is a
tyrosine-sulphated and widely distributed protein of the
extracellular matrixFEBS
Lett351433436199410.1016/0014-5793(94)00907-48082810
|
|
48.
|
A KatoO OkamotoK IshikawaH SumiyoshiN
MatsuoH YoshiokaM NomizuT ShimadaS FujiwaraDermatopontin interacts
with fibronectin, promotes fibronectin fibril formation, and
enhances cell adhesionJ Biol
Chem2861486114869201110.1074/jbc.M110.17976221398523
|
|
49.
|
B VidalAL SerranoM TjwaM SuelvesE ArditeR
De MoriB Baeza-RajaM Martinez de LagranP LafusteV
Ruiz-BonillaFibrinogen drives dystrophic muscle fibrosis via a
TGFbeta/alternative macrophage activation pathwayGenes
Dev2217471752200810.1101/gad.46590818593877
|
|
50.
|
M IshizakiT SugaE KimuraT ShiotaR KawanoY
UchidaK UchinoS YamashitaY MaedaM UchinoMdx respiratory impairment
following fibrosis of the diaphragmNeuromuscul
Disord18342348200810.1016/j.nmd.2008.02.00218358722
|