Introduction
Schwann cells (SCs), the principal supporting cells
of the peripheral nervous system, play a key role in peripheral
nerve regeneration. Autologous nerve grafting or biodegradable
conduits are the gold standard for peripheral nerve repair
(1). Use of primary somatic SCs
for peripheral nerve repair is limited because these cells are
difficult to prepare and maintain in high yield and purity under
common cell culture conditions.
Transfection of an exogenous hTERT, encoding the
catalytic subunit of human telomerase, can be used to prevent
telomere shortening, overcome telomere-controlled senescence, and
immortalize primary somatic cells. Most importantly, hTERT alone
can immortalize cells without causing cancer-associated changes or
altering phenotypic properties. Ectopic hTERT expression induces
immortalization of adult canine SCs (2) and primary human fetal SCs (3) without substantial alterations of
their phenotypes. Moreover, the nerve regeneration process is also
regulated by neurotrophic factors (NTFs) synthesized directly or
indirectly by SCs. Therefore, it is essential to evaluate the
secretion of NTFs after SCs are exposed to an exogenous hTERT.
However, there is limited reports on the relationship between the
secretion of NTFs and the transfection of hTERT.
In this study, we describe how the induction of the
hTERT gene allowed us to establish immortalized SCs derived from
rat embryo dorsal root ganglions. Additionally, we evaluated
whether these cell lines have SCs properties, and examined the
function of the secretion of NTFs (BDNF and NGF).
Materials and methods
Cell culture
A total of 30 healthy Sprague-Dawley female rats
were obtained by the Animal Experimental Center of Dalian Medical
University (License No. SCXK(Liao)2006-0002). Dorsal root ganglions
(DRG) from rat embryos were isolated under a microscope.
Non-nervous tissue was gently stripped under a dissecting
microscope, and the nerve segments were stored in ice-cold
Dulbecco’s modified Eagle’s medium (DMEM) containing 1%
penicillin/streptomycin. Briefly, dissected dorsal root ganglions
were digested with 0.3% collagenase type I solution (Sigma, USA)
and 0.25% trypsin (Sigma). After blocking the enzymatic digestion
with DMEM containing 10% fetal bovine serum (Gibco, USA), cells
were centrifuged and subsequently mechanically dissociated. At ∼75%
confluence after plating, cells were purified with 2 cycles of
cytosine arabinoside (10 mM) to eliminate fibroblasts and neurons
(4). Finally, the media was
changed with melanocyte growth medium (Cascade Biologics, Portland,
OR) to which 2 μM forskolin (Sigma), 10 ng/ml FGF-2 (Sigma) and 5
μg/ml bovine pituitary extract (Sigma) were added in order to
prevent fibroblast proliferation (5). All cultures were maintained at 37°C
in a humidified atmosphere of 5% CO2. The growth medium
was renewed every 2–3 days. SCs were subcultured to the next
passage when reaching 80–90% confluence.
Transfection of the hTERT gene
When SCs (passage 3) reached 90% confluence,
transfection was performed. SCs were transfected with pCI-neo-hTERT
plasmid (Yrbio, China) using Lipofectamine™2000 (Invitrogen, USA)
according to the manufacturer’s instructions. After selection with
400 μg/ml G418 (Sigma), the resistant clones were picked and
expanded under standard conditions with 100 μg/ml G418. The growth
medium was a melanocyte growth medium containing 2 μM forskolin
(Sigma), 10 ng/ml FGF-2 and 5 μg/ml bovine pituitary extract. SCs
were subcultured to the next passage when which on reaching 80–90%
confluence.
Indirect immunofluorescence staining
For immunocytochemistry, hTERT-SCs (passage 30)
cultured on coverslips were fixed with 4% paraformaldehyde (PFA) in
PBS for 10 min and then permeabilized with 0.05% Triton X-100 for
10 min. Non-specific sites were blocked with 5% goat serum for 1 h.
Then, the cells were incubated with primary antibodies for 12 h at
4°C. The following primary antibodies were used rabbit anti-S100β
(Boster, Wuhan, China), mouse anti-p75 (Boster), mouse antiglial
fibrillary acidic protein (anti-GFAP, Boster) all at 1:100
dilution. Blocking solution without a primary antibody was added
for 12 h at 4°C as a control. Antibody binding was detected by
using fluorescently labeled secondary antibodies (Vector
Laboratories, Burlingame, CA) at 1:200 dilution and cell nuclei
were counterstained with 4′,6-diamidino-2-phenylindole
dihydrochloride (DAPI,1:1000). The coverslips were then washed
three times in PBS (5 min each), and finally, the cells were
mounted in antifade solution (6).
Labeled cells were examined with fluorescence microscopy. The
images were digitally recorded and processed using Image-Pro
Plus.
Detection of telomerase activity
hTERT-SCs of passages 4, 8, 12, and 30 and SCs of
passage 3 were extracts corresponding to 104 cells, and
the telomerase activity was evaluated in these clones using the
TRAP assay according to the manufacturer’s instructions as
previously suggested (7). PCR
reaction products were separated on 12.5% non-denaturing acrylamide
gels. After fixation [0.5 M NaCl, 50% ethanol, and 40 mM sodium
acetate (pH 4.2)], the gels were directly exposed to an X-ray film
with an intensifying screen.
Reverse transcriptase polymerase chain
reaction (RT-PCR)
hTERT-SCs (5×104/ml) of passages 4, 8,
12, and 30 and SCs (5×104/ml) of passage 3 were
trypsinized and seeded onto 6-well plates. When cells reached 90%
confluence, RT-PCR was performed. Total RNA was harvested using
Trizol reagent (Invitrogen). Total RNA (200 ng) was reverse
transcribed for 45 min at 60°C, after which a two-step PCR
amplification was performed, according to the manufacturer’s
instructions. mRNA from cDNA samples was amplified with specific
primer pairs for β-actin, hTERT, NGF-β, BDNF, p16, and p53. The
primer sequences and the PCR conditions are depicted in Table I. Expression of hTERT, p53 and p16
in hTERT-SCs were examined at passage 4 and 30; HT-29 (HeLa,
ATCC:CCL-218) were used as positive controls. The secretion of NTFs
(BDNF and NGF) were examined at hTERT-SCs of passages 4, 8, 12 and
30, and untransfected SCs of passage 3 were used as control cells.
β-actin expression was detected as the internal control.
Amplification products were separated by 2.0% agarose gel
electrophoresis and visualized by ethidium bromide staining. After
the gels were scanned, the relative intensity of the bands was
determined by using the LabWorks 4.6 gel imaging and analysis
software (UVP, Inc.). The experiments were repeated three
times.
 | Table I.Primers used for RT-PCR. |
Table I.
Primers used for RT-PCR.
| Primer | Forward primer | Reverse primer | Ta | Amplicon length
(bp) |
|---|
| hTERT |
5′-TGTACTTTGTCAAGGTGGATGTG-3′ |
5′-GTACGGCTGGAGGTCTGTCAAG-3′ | 68°C | 200 |
| p16 |
5′-CTTCCTGGACACGCTGGT-3′ |
5′-CGAGGTACCGTGCGACAT-3′ | 60°C | 125 |
| p53 |
5′-GTTTCCGTCTGGGCTTCT-3′ |
5′-CCTCAGGCGGCTCATAG-3′ | 55°C | 351 |
| NGF-β |
5′-GGCCACTCTGAGGTGCATAG-3′ |
5′-CATGGGCCTGGAAGTCTAAA-3′ | 56°C | 349 |
| BDNF |
5′-AAACCATAAGGACGCGGACT-3′ |
5′-GATTGGGTAGTTCGGCATTG-3′ | 56°C | 393 |
| β-actin |
5′-TCTACGAGGGCTATGCTCTCC-3′ |
5′-GGATGCCACAGGATTCCATAC-3′ | 55°C | 320 |
Western blotting
To confirm the results from the secretions of NTFs
(BDNF and NGF), we detected the secretions of NTFs (BDNF and NGF)
at SCs of passages 4, 8, 12 and 30 after transfection respectively
using western blot analysis; untransfected SCs of passage 3 were
used as control cells. SCs were washed with PBS and lysed with a
lysis buffer (1% Triton X-100, 150 mM NaCl, 10 mM Tris, pH 7.4, 1
mM EDTA, 1 mM EGTA, pH 8.0, 0.2 mM Na3VO4,
0.2 mM PMSF, 0.5% Nonidet P-40) and incubated on ice for 30 min.
The cell lysates were then clarified by centrifugation at 9,000 x g
for 10 min at 4°C and the supernatant was saved for protein
analysis and western blotting. Total protein concentration was
determined by using a commercially available kit based on the
bicinchoninic acid (BCA) method. Total protein lysates (80 μg) were
separated by 10% SDS-PAGE mini-gel. Samples were transferred
electrophoretically to nitrocellulose membranes. The membrane was
blocked with 5% nonfat dry milk in Tris buffered saline with
Tween-20 (TTBS), washed with TTBS and incubated overnight with
anti-active NTFs (BDNF and NGF) (1:500) and β-actin antibody
(1:2000, Sigma). After washing in TTBS, the bound antibody was
detected through the use of avidin-conjugated horseradish
peroxidase (1:500, Sigma). Color reaction was obtained using
NBT/BCIP. The membranes were scanned and analyzed using the
LabWorks 4.6 software. The experiments were repeated three
times.
Statistical analysis
Results are expressed as mean ± SEM (standard error
of mean) of three independent experiments. One-way ANOVA followed
by the Newman Keuls multiple comparison test was used to compare
control and treated groups, with P<0.05 indicating significant
differences.
Results
Transfection of SCs, expression of hTERT,
p53 and p16
After the second purification procedure, the primary
SCs cultured within 2 weeks reached 90% confluence (Fig. 1A). Cells at early passage (passage
3) were transfected with the CI-neohTERT plasmid construct
expressing the catalytic subunit of the hTERT and a selection of
positive clones was conducted in G418 (100 μg/ml) containing
medium. Over 100 G418-resistant clones were isolated from each
experiment. However, the doubling time of hTERT-SCs was not
significantly modified after transfection (Fig. 1B).
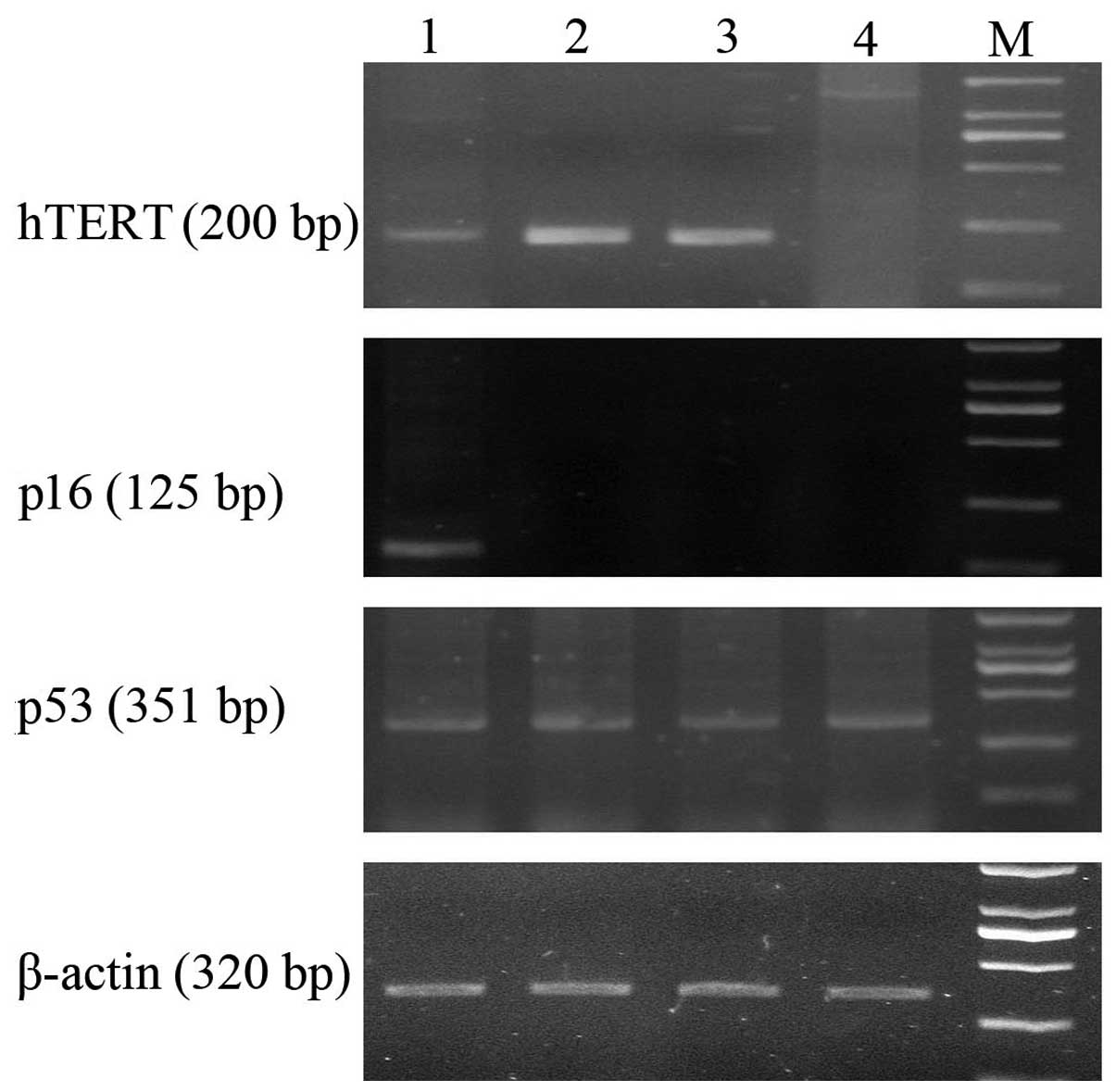
All the early (passage 4) and late (passage 30)
passages hTERT-SCs expressed hTERT mRNA. hTERT-SCs expressed p53
but lacked p16 as demonstrated by RT-PCR. In addition, p53 and p16
expression was not altered in early and late passages hTERT-SCs
compared to their non-transfected counterparts as demonstrated by
RT-PCR (Fig. 2).
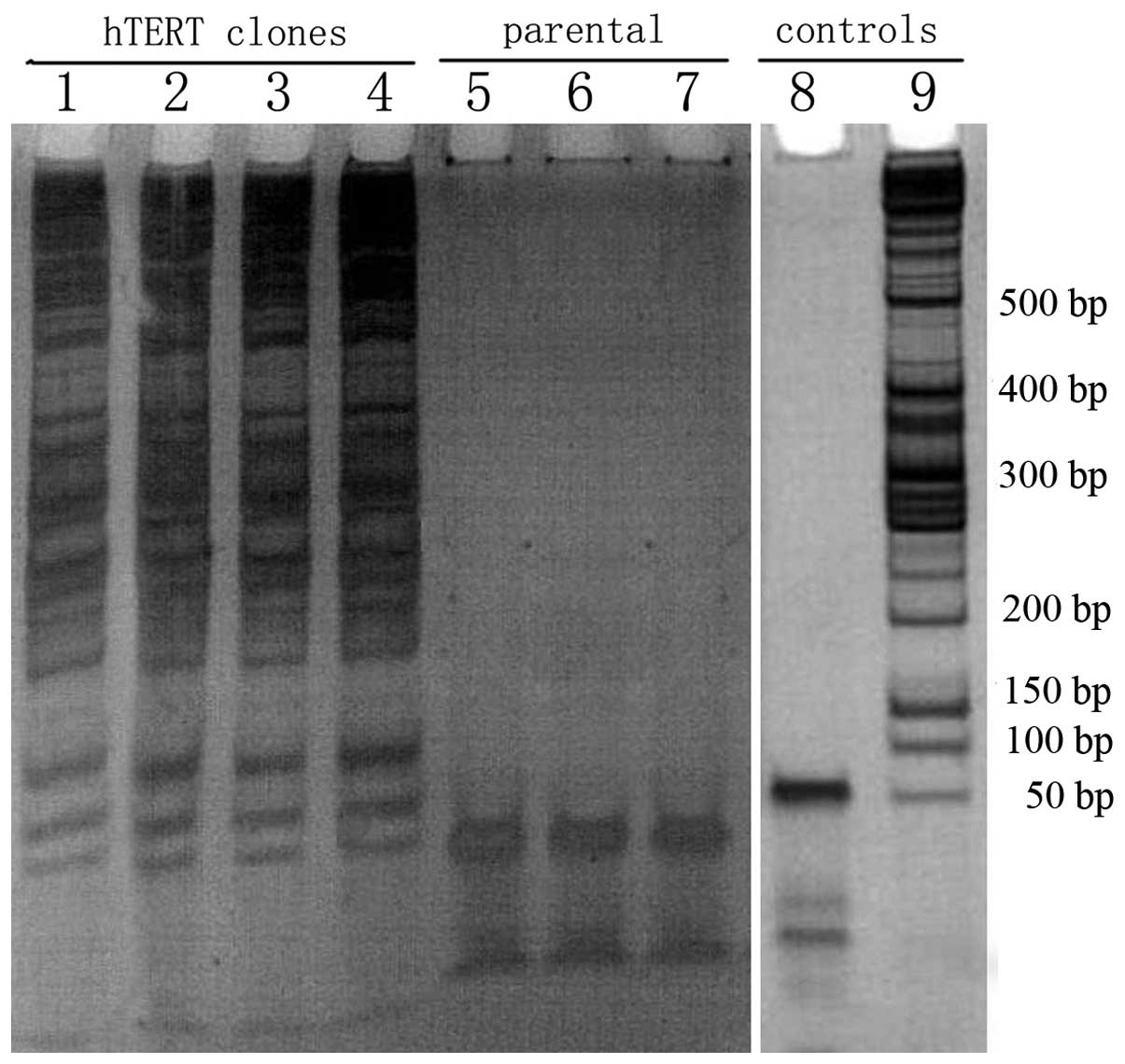
Telomerase activity
hTERT-SCs were expanded from each passage (passages
4, 8, 12 and 30) and the telomerase activity was detected in these
clones using the TRAP assay. Telomerase activity was detected in
all hTERT-SCs passages and these cells displayed full telomerase
activity (Fig. 3). This activity
was absent in control clones (parental non-transfected cells).
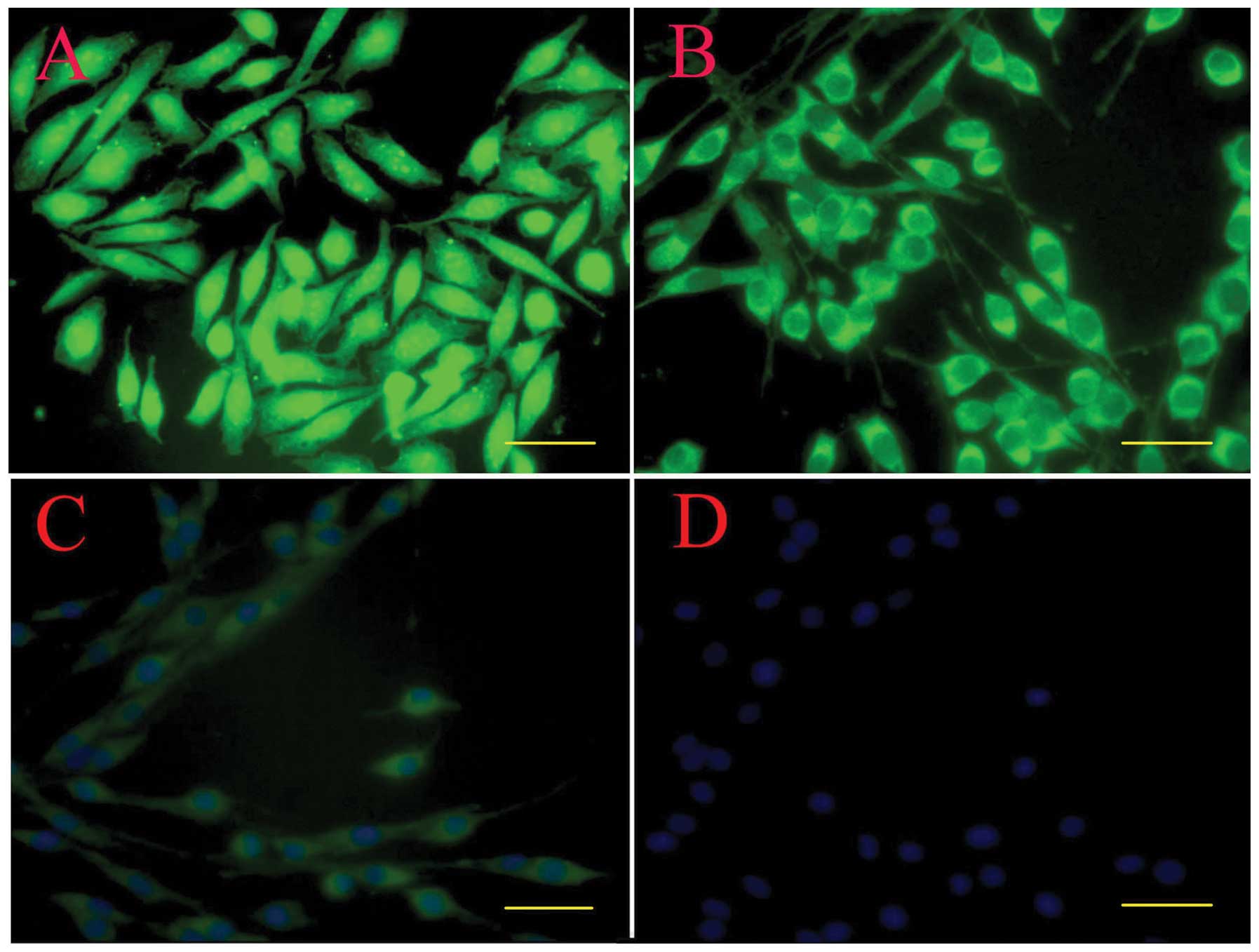
Immunostaining
By immunostaining, hTERT-SCs at passage 30 still
showed a positive staining for GFAP, p75 and S100β. The staining
was evident in the cell body and along the processes. These cells
demonstrated typical bipolar or tripolar morphology in
vitro, brightly stained for p75 (Fig. 4A), GFAP (Fig. 4B) and S100β (Fig. 4C), and had oval nuclei (Fig. 4D).
NGF and BDNF mRNA expression detected by
RT-PCR
We studied the effect of hTERT on the biological
activities of SCs by examining NGF and BDNF mRNA through the
expression RT-PCR. A semi-quantitative RT-PCR was adopted to
analyze the expression of NGF and BDNF mRNA (Fig. 5). The relative density of a 349 bp
product of the NGF gene, a 393 bp product of the BDNF gene
respectively and a 320 bp product of the internal control β-actin
was calculated after separation by 1% agarose gel electrophoresis.
Results showed that the expression of the BDNF gene in the
transfected SCs was dramatically decreased as cell passage
increased, compared to the untransfected control, while the
expression of NGF was elevated at early passages (4 and 8) and
decreased at late passages (12 and 30). The alteration of gene
expression of NGF (Fig. 5B) and
BDNF (Fig. 5C) in the transfected
SCs was passage-dependent.
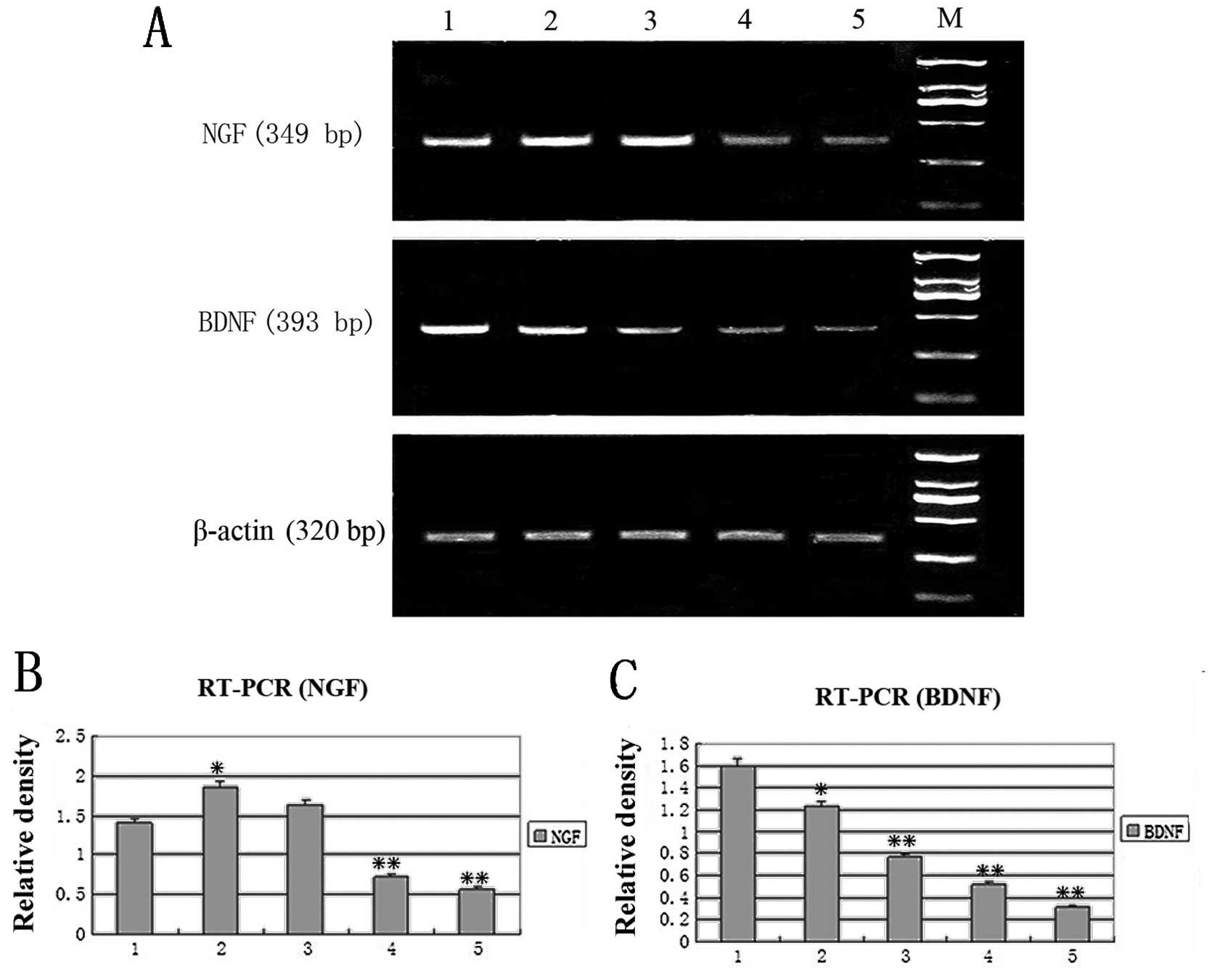 | Figure 5.RT-PCR analysis of NGF and BDNF. (A)
Lane 1, untransfected control cells. Lanes 2–5, hTERT-SCs of
passages 4, 8, 12 and 30, respectively. M, marker. The relative
density of (B) NGF and (C) BDNF mRNA vs. β-actin mRNA
respectively(*P<0.05,**P<0.01). Lane 1,
untransfected control cells. Lanes 2–5, hTERT-SCs of passages 4, 8,
12 and 30, respectively. M, marker. |
NGF and BDNF protein expression detected
by western blotting
To detect the protein expression level of BDNF and
NGF in the transfected SCs, western blotting was employed (Fig. 6). Results revealed that the
expression level of BDNF BDNF gene in the transfected SCs was
dramatically decreased as cell passage increased, compared to the
untransfected control, while the expression level of NGF was
elevated at early passages (4 and 8) and decreased at late passages
(12 and 30). In Fig. 6, western
blot analysis further confirmed that the alteration of gene
expression of NGF (Fig. 6B) and
BDNF (Fig. 6C) in the transfected
SCs was passage-dependent.
 | Figure 6.Western blot analysis of NGF and
BDNF. Lane 1, untransfected control cells. Lanes 2–5, hTERT-SCs of
passage 4, 8, 12 and 30 respectively. The relative density of (B)
NGF and (C) BDNF mRNA vs. β-actin mRNA
respectively(*P<0.05,**P<0.01). Lane 1,
untransfected control cells. Lanes 2–5, hTERT-SCs of passages 4, 8,
12 and 30 respectively. M, marker. |
Discussion
Schwann cells (SCs) are neural crest derivatives
that ensheathe and myelinate axons of peripheral nerves. They wrap
individually around the shaft of peripheral axons, forming myelin
sheaths along segments of the axon. SCs play an important role in
the development, function and regeneration of peripheral nerves.
They can enhance both peripheral and central nerve regeneration by
providing a supportive environment for neurite outgrowth through
the release of neurotrophic factors (8–10)
and cellular matrix protein (11,12). In addition, SCs promote axonal
regeneration and remyelination following transplantation into the
lesioned nervous system (13,14). Compared to the use of a conduit
alone, artificial nerve grafts containing SCs is a promising method
for peripheral nerve repair (15). However, for the application of SCs
in tissue engineering or for clinical use, the production of a
large number of viable SCs is necessary. Expansion and maintenance
of SCs in culture have been difficult, due to the low division rate
and potential overgrowth of fibroblasts over time (16–18).
We have demonstrated how the transfer of an
exogenous hTERT, encoding the catalytic subunit of human
telomerase, can be used to prevent telomere shortening, overcome
telomere-controlled senescence, and immortalize primary somatic
cells. The introduction of the hTERT gene into primary somatic
cells results in telomere length elongation and in the extension of
the in vitro replicative lifespan of primary somatic cells,
then increase cellular proliferation. The major advantage of using
hTERT exclusively to immortalize primary somatic cells is that the
enzyme telomerase can immortalize without causing cancer-associated
changes or altering phenotypic properties (19–21). Adult canine Schwann cells SCs
(2) and primary human fetal SCs
(3) have been immortalized by
hTERT or/and SV40 large T antigen.
In the present study, we used human telomerase
reverse transcriptase (hTERT) transfection of postnatal SD rats
Schwann cells derived from DRG to establish a stable source for
cell transplantation. The morphological phenotypes of transfected
SCs were not altered. All the early (passage 4) and late (passage
30) passage hTERT-SCs expressed hTERT mRNA and gained full
telomerase activity. This may indicate that SCs were successfully
transfected. By immunostaining, the hTERT-SCs at passage 30 still
showed a positive staining for S100β, p75, and GFAP without
altering phenotypic properties. This indicates that the transfected
SCs retained the properties of a primary SCs yet they were easy to
grow and propagate. In addition, p53 and p16 expression was not
altered in early and late passage transfected SCs compared to their
non-transfected counterparts. This may indicate that rat SCs
undergo senescence through a telomere-dependent pathway similar to
human cells as previously described (22,23).
Many studies introduced hTERT at a time when
telomeres had reached a critical length and hTERT expression
enhanced cellular proliferation (24–27). One important feature of SCs as a
promising cell type for transplantation is their ability to produce
a variety of trophic factors that are growth-promotive (28). It was demonstrated that SCs
expressed NGF (29), and BDNF
(30), and NT-3 (31). Among these factors, members of the
neurotrophin family play particular roles. However, little is known
about the effects of hTERT treatment on the secretion of SCs.
Based on our results hTERT increased the mRNA level
of NGF at early passage, whereas an adverse effect occurred on the
mRNA levels and protein secretion of BDNF in SCs at early passages
(4 and 8). These results indicated that hTERT is able to enhance
the biological activities of SCs through different effects on NGF
and BDNF, and hTERT induced different mechanisms responsible for
regulating NGF and BDNF expression. This study is similar to the
effect of Hypoxia/Reoxygenation on SCs (32), which demonstrated that the
time-course and spatial pattern of both expression are distinctly
different. After transfection, early cell passages that increased
NGF may be correlated with protecting SCs, and stimulating SCs
retrodifferentiation and proliferation. However, the function of
BDNF, such as inducing SCs and expression of myeline proteins, is
not similar to NGF (33). SCs
exposed to hTERT, upregulated NGF to meet an urgent requirement of
protecting SCs, and downregulated BDNF, a non-urgent requirement. A
decrease in BDNF and subsequent decrease in NGF may be correlated
with either hTERT-SCs secretion decrease or the decrease in the
number of cells.
Conventional methods for extending cellular lifespan
almost invariably alter the phenotypic properties of cells, thereby
reducing the value of the immortalized cells. However, the levels
of BDNF were decreased at all passages and those of only NGF at
late passages (12 and 30), demonstrating that telomerase can
stimulate the proliferation and secretion of SCs at early passages,
but does not lead to their immortalization. This study also
demonstrates that the ability of proliferation and secretion of
hTERT-SCs will decrease when they reach a specific point in
time.
In summary, we successfully transfected SCs with
hTERT and observed that these cells displayed full telomerase
activity. Differences in the expression of p53 and p16 prior to and
after transfection were not detected. However hTERT alone is not
always sufficient to immortalize primary somatic cells. This method
can extend the lifespan of the cells without causing
cancer-associated changes or alter phenotypic properties.
Acknowledgements
This study was supported by the
National Natural Science Foundation of China, no. 30973060.
References
|
1.
|
SE MackinnonAL DellonA study of nerve
regeneration across synthetic (Maxon) and biologic (collagen) nerve
conduits for nerve gaps up to 5 cm in the primateJ Reconstr
Microsurg6117121199010.1055/s-2007-10068102352218
|
|
2.
|
S TechangamsuwanR KreutzerM KreutzerI
ImbschweilerK RohnK WewetzerW BaumgärtnerTransfection of adult
canine Schwann cells and olfactory ensheathing cells at early and
late passage with human TERT differentially affects growth factor
responsiveness and in vitro growthJ Neurosci
Methods176112120200910.1016/j.jneumeth.2008.08.030
|
|
3.
|
HC LehmannW ChenR MiS WangY LiuM RaoA
HökeHuman Schwann cells retain essential phenotype characteristics
after immortalizationStem Cells
Dev21423431201210.1089/scd.2010.0513
|
|
4.
|
Y WeiJ ZhouZ ZhengA WangQ AoY GongX
ZhangAn improved method for isolating Schwann cells from postnatal
rat sciatic nervesCell Tissue
Res337361369200910.1007/s00441-009-0836-419639342
|
|
5.
|
A NiapourF KaramaliK KarbalaieA KianiM
MardaniMH Nasr-EsfahaniH BaharvandNovel method to obtain highly
enriched cultures of adult rat Schwann cellsBiotechnol
Lett32781786201010.1007/s10529-010-0230-z20213527
|
|
6.
|
T KomiyamaY NakaoY ToyamaH AsouCA
VacantiMP VacantiA novel technique to isolate adult Schwann cells
for an artificial nerve conduitJ Neurosci
Methods122195200200310.1016/S0165-0270(02)00320-512573478
|
|
7.
|
WE WrightJW ShayMA PiatyszekModifications
of a telomeric repeat amplification protocol (TRAP) result in
increased reliability, linearity and sensitivityNucleic Acids
Res2337943795199510.1093/nar/23.18.3794
|
|
8.
|
JW FawcettRJ KeynesPeripheral nerve
regenerationAnnu Rev
Neurosci134360199010.1146/annurev.neuro.13.1.43
|
|
9.
|
MR FeneleyJW FawcettRJ KeynesThe role of
Schwann cells in the regeneration of peripheral nerve axons through
muscle basal lamina graftsExp
Neurol114275285199110.1016/0014-4886(91)90153-41748202
|
|
10.
|
K HaastertJ GrosskreutzM JaeckelC LadererJ
BuflerC GrotheP ClausRat embryonic motoneurons in long-term
co-culture with Schwann cells - a system to investigate motoneuron
diseases on a cellular level in vitroJ Neurosci
Methods142275284200510.1016/j.jneumeth.2004.09.003
|
|
11.
|
K RothblumRC StahlDJ CareyConstitutive
release of alpha4 type V collagen N-terminal domain by Schwann
cells and binding to cell surface and extracellular matrix heparan
sulfate proteoglycansJ Biol
Chem2795128251288200410.1074/jbc.M408837200
|
|
12.
|
Y ShibuyaA MizoguchiM TakeichiK ShimadaC
IdeLocalization of N-cadherin in the normal and regenerating nerve
fibers of the chicken peripheral nervous
systemNeuroscience67253261199510.1016/0306-4522(95)00015-B7477906
|
|
13.
|
A Baron-Van EvercoorenV Avellana-AdalidF
LachapelleR LiblauSchwann cell transplantation and myelin repair of
the CNSMult Scler315716119979291173
|
|
14.
|
YY ChenD McDonaldC ChengB MagnowskiJ
DurandDW ZochodneAxon and Schwann cell partnership during nerve
regrowthJ Neuropathol Exp
Neurol64613622200510.1097/01.jnen.0000171650.94341.4616042313
|
|
15.
|
A MosahebiB WoodwardM WibergR MartinG
TerenghiRetroviral labeling of Schwann cells: in vitro
characterization and in vivo transplantation to improve peripheral
nerve regenerationGlia34817200110.1002/glia.103511284015
|
|
16.
|
JL RutkowskiCJ KirkMA LernerGI
TennekoonPurification and expansion of human Schwann cells in
vitroNat Med18083199510.1038/nm0195-807584959
|
|
17.
|
TK MorrisseyN KleitmanRP BungeHuman
Schwann cells in vitro. II. Myelination of sensory axons following
extensive purification and heregulin-induced expansionJ
Neurobiol28190201199510.1002/neu.480280206
|
|
18.
|
TK MorrisseyRP BungeN KleitmanHuman
Schwann cells in vitro. I Failure to differentiate and support
neuronal health under co-culture conditions that promote full
function of rodent cellsJ
Neurobiol28171189199510.1002/neu.480280205
|
|
19.
|
XR JiangG JimenezE ChangM FrolkisB KuslerM
SageM BeecheAG BodnarGM WahlTD TlstyCP ChiuTelomerase expression in
human somatic cells does not induce changes associated with a
transformed phenotypeNat Genet21111114199910.1038/50569916802
|
|
20.
|
CP MoralesSE HoltM OuelletteKJ KaurY YanKS
WilsonMA WhiteWE WrightJW ShayAbsence of cancer-associated changes
in human fibroblasts immortalized with telomeraseNat
Genet21115118199910.1038/50639916803
|
|
21.
|
MM OuelletteLD McDanielWE WrightJW ShayRA
SchultzThe establishment of telomerase-immortalized cell lines
representing human chromosome instability syndromesHum Mol
Genet9403411200010.1093/hmg/9.3.40310655550
|
|
22.
|
DJ ArgyleL NasirTelomerase: a potential
diagnostic and therapeutic tool in canine oncologyVet
Pathol4017200310.1354/vp.40-1-112627707
|
|
23.
|
L NasirTelomeres and telomerase:
Biological and clinical importance in dogsVet
J175155163200810.1016/j.tvjl.2007.01.02417398127
|
|
24.
|
V GorbunovaA SeluanovOM
Pereira-SmithEvidence that high telomerase activity may induce a
senescent-like growth arrest in human fibroblastsJ Biol
Chem27876927698200310.1074/jbc.M21294420012496279
|
|
25.
|
LL SmithHA CollerJM RobertsTelomerase
modulates expression of growth-controlling genes and enhances cell
proliferationNat Cell Biol5474479200310.1038/ncb98512717449
|
|
26.
|
JI YoungJM SedivyJR SmithTelomerase
expression in normal human fibroblasts stabilizes DNA
5-methylcytosine transferase IJ Biol
Chem2781990419908200310.1074/jbc.M30168520012665523
|
|
27.
|
X JinJS LeeS KwakSY LeeJE JungTK KimC XuZ
HongZ LiSM KimEstablishment and characterization of three immortal
bovine muscular epithelial cell linesMol
Cells212933200616511344
|
|
28.
|
M OudegaXM XuSchwann cell transplantation
for repair of the adult spinal cordJ
Neurotrauma23453467200610.1089/neu.2006.23.45316629629
|
|
29.
|
R HeumannS KorschingC BandtlowH
ThoenenChanges of nerve growth factor synthesis in nonneuronal
cells in response to sciatic nerve transectionJ Cell
Biol10416231631198710.1083/jcb.104.6.16233034917
|
|
30.
|
A AchesonPA BarkerRF AldersonFD MillerRA
MurphyDetection of brain-derived neurotrophic factor-like activity
in fibroblasts and Schwann cells: inhibition by antibodies to
NGFNeuron7265275199110.1016/0896-6273(91)90265-21873030
|
|
31.
|
N OffenhauserR Böhm-MatthaeiP TsoulfasL
ParadaM MeyerDevelopmental regulation of full-length trkC in the
rat sciatic nerveEur J
Neurosci7917925199510.1111/j.1460-9568.1995.tb01079.x7613627
|
|
32.
|
H ZhuF LiWJ YuWJ WangL LiLD WanY LeWL
DingEffect of hypoxia/reoxygenation on cell viability and
expression and secretion of neurotrophic factors (NTFs) in primary
cultured schwann cellsAnat Rec
(Hoboken)293865870201010.1002/ar.2110520186961
|
|
33.
|
N OkaT KawasakiK MizutaniH SugiyamaI
AkiguchiHypoxia-inducible factor 1alpha may be a marker for
vasculitic
neuropathyNeuropathology27509515200710.1111/j.1440-1789.2007.00817.x18021370
|