Introduction
Hyaline cartilage is characterised by avascularity,
a rich extracellular matrix (ECM), low cell density and turnover.
As a consequence, the capacity of cartilage regeneration is
considerably reduced compared to other connective tissues. As a
result, traumatic cartilage defects (1) and degenerative changes as a
consequence of the rising age (2)
are associated with joint pain and limited functions (3) and ultimately lead to osteoarthritis.
Therefore, the major challenge of a successful treatment of
cartilage defects in orthopaedic surgery is to prevent this
progressive degeneration.
Autologous chondrocyte transplantation (ACT) is a
possible technique for cartilage treatment with promising results
(4). For this method, the
patient’s own chondrocytes need to be isolated and afterward
cultivated in vitro to increase cell number. Hereby,
proliferation of cells is associated with a de-differentiation
process towards a fibroblastoid phenotype. As a result,
chondrocytes lose their differentiated characteristics like
collagen type 2 and proteoglycan synthesis (5). In order to synthesise hyaline ECM
components, cells have to re-differentiate into the chondrocytic
phenotype. For cartilage regeneration, it is well known that a
three-dimensional (3D) environment could support re-differentiation
processes of chondrocytes (6,7).
Furthermore, essential chondrogenic growth factors were identified
which could also support differentiation processes in vitro.
Hereby, growth factors are stored in the ECM and will be released
under specific conditions (e.g. surface damaging) to regulate
cellular behaviour (8).
One important growth factor in cartilage is the
insulin-like growth factor (IGF)-1 which is the major anabolic
factor for stimulating matrix synthesis by working in an autocrine
and paracrine way (9). IGF-1
effects and therefore the biological function are modulated by the
interaction of IGF-binding proteins (IGFBP). Due to the binding of
IGF-1 to IGFBP, the transport and also the availability of IGF-1 to
chondrocytes are regulated (8).
Beside IGF-1, transforming growth factor (TGF)-β1 is
also responsible for matrix synthesis by increasing the expression
of collagen type 2 and accumulating proteoglycans (10). However, effects of TGF-β1 on
chondrocytes are dependent on the conditions of the target cells.
TGF-β1 could stimulate as well as inhibit cell proliferation and
proteoglycan synthesis (11). It
is well described that the combination of TGF-β1 and IGF-1 causes
an induction of collagen type 2 mRNA in de-differentiated
chondrocytes derived from articular cartilage (12). Furthermore, in
alginate-encapsulated de-differentiated chondrocytes the treatment
of IGF-1 and TGF-β1 resulted in an accumulation of ECM components
like collagen type 2, glycosaminoglycan (GAG) and aggrecan
(13,14).
The aim of this study was to analyse the effects of
TGF-β1, IGF-1 and the combination of both on the proliferation and
differentiation capacity of human chondrocytes in pellet cultures
under different oxygen concentrations. Hereby, we investigated
which conditions could have a positive effect on chondrogenic
re-differentiation and therefore accumulation of ECM.
For the experiments we used cells derived from
osteoarthritic (OA) human cartilage which is characterised by
limited reactions onto IGF-1 and TGF-β (15). One possible reason could be the
increased oxidative stress and therefore the enrichment of reactive
oxygen species (ROS) which lead to a reduced effect of IGF-1
(2). Therefore, we hypothesise
that the reduction of the oxygen content from 21% (normoxia) to 5%
(hypoxia) during cultivation could influence the effects of IGF-1
and TGF-β1 positively because of a decreased oxidative stress for
cells.
Materials und methods
Chondrocyte isolation and
cultivation
Articular knee cartilage was obtained from 12
patients (4 male donors, mean age, 71±12.4 years; 8 female donors,
mean age, 69.8±6.8 years) undergoing primary knee replacement. The
samples were collected after obtaining consent from the patients
and approval by the Local Ethics Committee (registration no. A2009
17).
For isolation of primary chondrocytes the cartilage
was removed from the underlying bone, cut into small pieces and
washed three times in PBS (PAA Laboratories, Coelbe, Germany).
Afterwards the cartilage (∼3 g w/w) was treated with 1%
Gibco® Trypsin/EDTA (Invitrogen, Darmstadt, Germany) for
20 min at 37°C followed by 0.2% collagenase A (Roche Diagnostics
GmbH, Mannheim, Germany) in DMEM supplemented with 10% FCS, 1%
amphotericin B and 1% Gibco® penicillin/streptomycin
(Invitrogen) at 37°C overnight. The cell suspension was filtered
through a cell strainer (pore size, 70 μm; Nunc, Wiesbaden,
Germany) and centrifuged at 118 x g for 10 min. The cell pellet was
resuspended in DMEM with the supplements mentioned above and
ascorbic acid.
Freshly isolated chondrocytes were plated in a
25-cm2 culture flask with 8 ml culture medium with
supplements and incubated in a humidified atmosphere at 37°C and 5%
CO2. The medium was changed every second day. Cell
proliferation was determined visually by microscopic control. When
cell confluence reached 90% (∼5×105 cells/25
cm2 flask), the cells were trypsinised and split at a
ratio 1 to 6. For all experiments cryo-conserved chondrocytes were
used. After thawing, cells were centrifugated at 118 x g for 10
min, transferred into 75-cm2 flasks (passage 2) and
incubated in a humidified atmosphere at 37°C, 5% CO2 and
5% O2 (hypoxia) or 21% O2 (normoxia).
In passage three, chondrocyte pellet cultures with
an amount of 5×105 cells/pellet were prepared.
Therefore, cells were trypsinised, centrifuged at 118 × g for 10
min and an appropriate aliquot of cells was split in three
different medium compositions. Subsequently, the cell suspension
was transferred into conical tubes. Pellet cultures were incubated
in serum-free DMEM containing ascorbic acid (final concentration,
50 μg/ml; Sigma) dexamethasone (final concentration, 100 nM;
Sigma), ITS™ (complete medium to ITS™ in a 100:1 ratio;
Becton-Dickinson, Heidelberg, Germany) and the chondrogenic growth
factors IGF-1 (R&D Systems, Wiesbaden, Germany) and/or TGF-β1
(Tebu-Bio, Offenbach, Germany) in three different compositions,
shown in Table I, at 37°C in a
humidified atmosphere of 5% CO2 and the appropriate
oxygen concentration for 5 weeks. The culture medium was also
changed every second day.
 | Table I.Three different growth factor
combinations in the cell medium. |
Table I.
Three different growth factor
combinations in the cell medium.
| Medium combination
1 | Medium combination
2 | Medium combination
3 |
|---|
| TGF-β1 | 50 ng/ml | 50 ng/ml | - |
| IGF-1 | 50 ng/ml | - | 50 ng/ml |
DNA isolation and quantification
For the DNA isolation pellet cultures of each
stimulating experiment were transferred in 2 ml homogenisation
tubes containing small steel beads (Precellys-Steel kit, 2.8 mm;
PeqLab Biotechnologie GmbH, Erlangen, Germany), covered with 100 μl
TE-buffer and homogenised for 30 sec at 5,000 x g.
DNA isolation was performed using the peqGOLD Tissue
DNA mini kit (PeqLab Biotechnologie GmbH) according to the
manufacturer’s instructions. Afterwards, DNA concentrations were
measured with the Qubit Fluorometer according to the manufacturer’s
instructions (Invitrogen).
Pro-collagen type 1 (CICP) and type 2
(CP2) quantification
Synthesis of pro-collagen type 1 (Quidel, Marburg,
Germany) and type 2 (TECOmedical, Buende, Germany) were determined
by ELISA. Hence, supernatants of every medium combination were
collected and stored at −20°C after 14 and 35 days of incubation.
The assay was performed according to manufacturer’s instructions.
Absorbance was measured at 450 nm for the CP2 ELISA and 405 nm for
the CICP ELISA using Opsys MR™ microplate reader (Dynex
Technologies, Denkendorf, Germany). The content of pro-collagen
type 1 as well as type 2 was normalised to the DNA content (ng
pro-collagen/ng DNA).
Aggrecan (PG-EASIA) quantification
The PG-EASIA (Invitrogen) is a solid phase enzyme
amplified sensitivity immunoassay (EASIA). The background of this
assay is the use of an oligoclonal system in which monoclonal
antibodies are directed against distinct epitopes of
proteoglycan.
After 14 or 35 days of incubation, supernatants of
every medium combination were collected and stored at −20°C. The
assay was performed according to manufacturer’s instructions.
Absorbance was measured at 450 nm using the Opsys MR™ microplate
reader (Dynex Technologies). The content of aggrecan was normalised
to the DNA content (ng aggrecan/ng DNA).
Immunohistochemistry of pellet
cultures
For immunohistological examinations of type 2
collagen and aggrecan after 35 days of incubation, pellets were
fixed in 4% formaline (Merck, Darmstadt, Germany), embedded in
paraffin and sectioned to 7 μm slices.
For immunohistochemistry of collagen type 2, pellet
cultures were digested with pronase (Roche) in PBS buffer and
hyaluronidase (Sigma) in PBS buffer containing 0.01% BSA at 37°C
for 30 min in each case to increase the penetration of antibodies
into the matrix. Before incubation with the primary antibody, beads
were washed in PBS buffer for 5 min and blocked in PBS buffer
containing 5% goat serum (PAN Biotech, Aidenbach, Germany) for 30
min at room temperature. Mouse anti-human type 2 collagen antibody
(Millipore, Schwalbach/Ts., Germany) was applied at 1:200 dilution
in PBS buffer containing 0.1% goat serum overnight at 4°C. To
detect collagen type 2, Alexa Fluor® 488 goat anti-mouse
IgG (Invitrogen), diluted 1:200 in PBS buffer, was used for 2 h at
room temperature.
For aggrecan staining, slices of pellet cultures
were digested with chondroitinase (Sigma) in 20 mM Tris buffer
(Merck) and keratanase (Sigma) in 50 mM Tris buffer at 37°C and 5%
CO2 for 30 min. Afterwards, beads were washed in PBS
buffer for 5 min and blocked in PBS buffer containing 5% goat
serum. The monoclonal anti-human aggrecan G1-IGD-G2 domain antibody
(R&D Systems), diluted 1:200 in PBS with 1% goat serum, was
incubated overnight at 4°C. For aggrecan detection, Alexa
Fluor® 488 goat anti-mouse IgG, diluted 1:200 in PBS
containing 1% goat serum was applied for 1 h at room temperature.
For negative controls, the primary antibody was replaced by PBS
buffer and secondary antibody alone.
Afterwards, sections were embedded in Fluoroshield
mounting medium containing DAPI (Acris Antibodies GmbH, Herford,
Germany) to visualise cell nuclei. Evaluation and documentation of
the results were performed using a Nikon Eclipse TS100 microscope
with a Nikon Digital Sight DS-2Mv camera. Photographs were acquired
and processed with the NIS Elements 3.0 software (Nikon GmbH,
Düsseldorf, Germany).
Statistical analysis
Data are presented as means ± standard deviation.
For all analysis human chondrocyte cultures of a minimum of three
independent donors were used. Statistical significance between the
two time points was calculated with the Wilcoxon test. Comparison
between the three medium combinations was calculated with the
Kruskal-Wallis test for independent samples or the Mann-Whitney
test for paired samples. Comparison between both culture conditions
was calculated with the Mann-Whitney test. All statistical analyses
were performed with SPSS 15.0 for Windows (SPSS Inc., Chicago, IL,
USA). The significance level was determined for P<0.05.
Results
Chondrocyte pellet culture morphology and
proliferation
During incubation of human chondrocytes over a
period of 35 days under different growth factor combinations and
oxygen concentrations the cell pellets stimulated with IGF-1 showed
a compact appearance. This was in contrast to TGF-β1/IGF-1 and
TGF-β1 treated cultures, i.e. both pellet cultures were
approximately twice as large and showed a rounded to oval
appearance. Furthermore, under normoxic conditions chondrocytes of
these stimulation experiments showed a higher metabolic activity
than cells incubated in IGF-1 alone which was visible by a clear
colour change of the medium as a result of acidification. In
contrast, under hypoxia pellets of all three stimulating
experiments showed an enhanced metabolism during the incubation
time.
Under hypoxic conditions DNA contents of all three
stimulating experiments remained stable. Cell pellets revealed
nearly the same concentration of DNA at both time points after
stimulation with TGF-β1/IGF-1 and with TGF-β1. In IGF-1 stimulated
pellets a reduced DNA content was observable at both time points
compared to the other stimulating experiments. In contrast,
normoxic cultivated cells showed an enhanced proliferation after
stimulation with TGF-β1/IGF-1 and TGF-β1. Nevertheless, DNA
contents under normoxia did not reach the values of
hypoxic-cultivated cells. For IGF-1 stimulated pellet cultures a
decreased proliferation rate could be observed from Day 14 to 35
(Fig. 1).
Expression of pro-collagen type 1
Synthesis of pro-collagen type 1 declined during the
incubation time under hypoxic as well as under normoxic cell
culture conditions. Additionally, no significant differences could
be determined between both culture conditions. Under hypoxic
conditions similar synthesis rates for TGF-β1/IGF-1- as well as
TGF-β1-stimulation (Fig. 2) after
14 and 35 days were observable. After IGF-1 stimulation, reduced
contents of pro-collagen type 1 were determined compared to the
other stimulating experiments. Under normoxia the expression was
significantly reduced (P=0.04) on Day 14 compared to
TGF-β1/IGF-1-treated cells. Nevertheless, after 35 days of
incubation significant differences between IGF-1 and the other
growth factor combinations were visible for both culture conditions
(hypoxia, P=0.029; normoxia, P=0.002).
Expression of pro-collagen type 2
Expression of pro-collagen type 2 revealed
significant differences between hypoxia and normoxia at both time
points [TGF-β1/IGF-1, P= 0.017 (Day 14); TGF-β1, P= 0.03 (Day 14),
P= 0.004 (Day 35); IGF-1, P=0.017 (Day 14), P=0.004 (Day 35)].
After 14 days of stimulation, hypoxically cultivated cells showed a
decreased but similar type 2 pro-collagen content after treatment
in all stimulating experiments compared to normoxic cultivated
cells (Fig. 3). On Day 35, a
significantly (P=0.043) increased synthesis rate of pro-collagen
type 2 could be determined after TGF-β1/IGF-1 stimulation under
hypoxia. In contrast, the content of pro-collagen type 2 remained
stable after sole stimulation with TGF-β1 or IGF-1.
Under normoxic culture conditions significant
differences between IGF-1 and TGF-β1/IGF-1 (P=0.026) as well as
IGF-1 and TGF-β1 (P=0.002) stimulated pellets were detected after
35 days. Additionally, an increased synthesis rate could only be
shown after single treatment with TGF-β1 (Fig. 3).
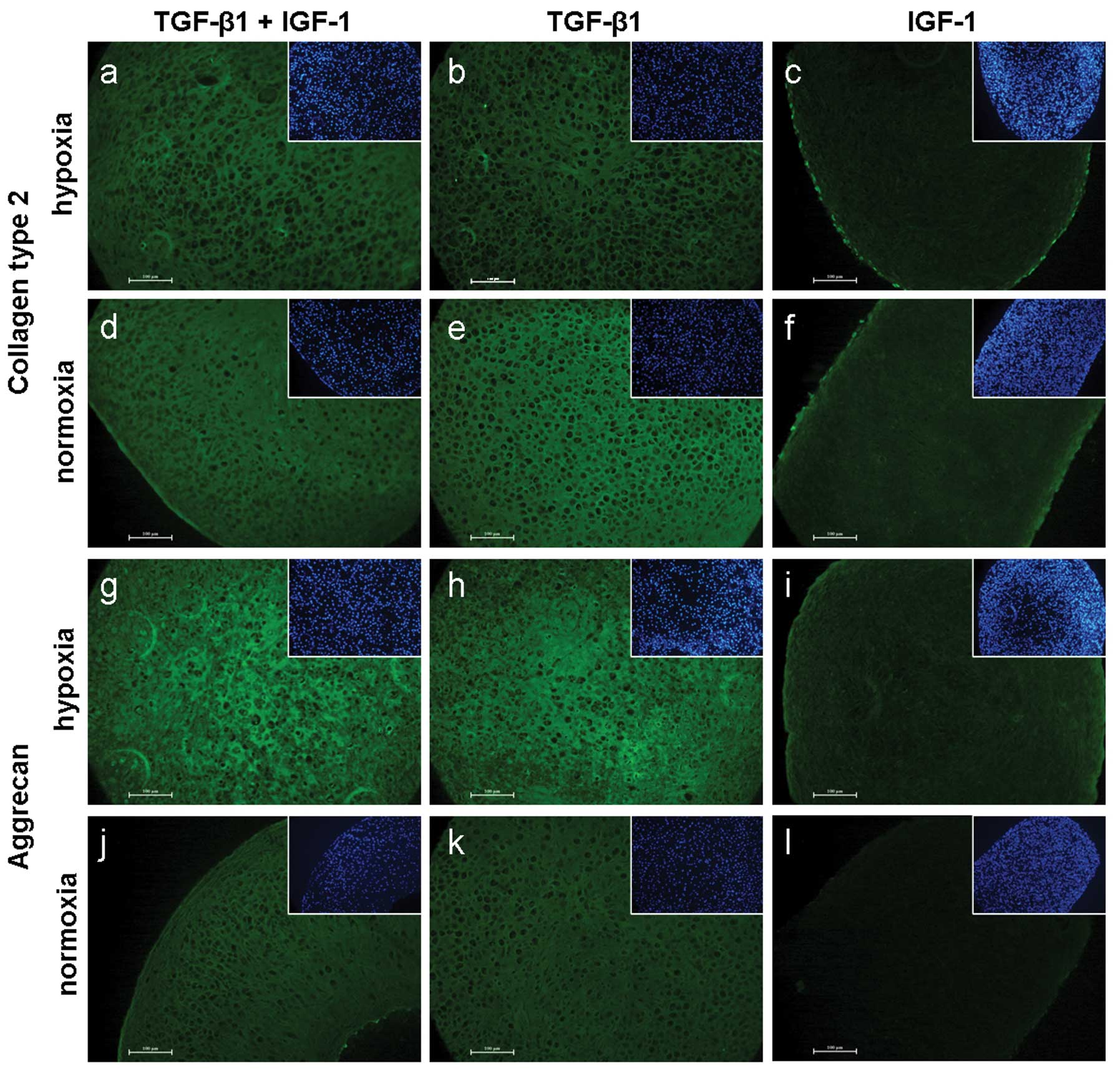
Collagen type 2 staining resulted in marginal
differences between hypoxia and normoxia. Compared to TGF-β1/IGF-1
and TGF-β1 treatments, IGF-1 stimulated pellets showed a less
pronounced staining under both culture conditions (Fig. 4a–f).
Synthesis of aggrecan
Under hypoxic culture conditions the synthesis of
aggrecan resulted in a distinct but non-significant accumulation of
the protein content from Day 14 to 35. At both time points,
significant differences could be determined between IGF-1
stimulated cells and TGF-β1/IGF-1 as well as TGF-β1 stimulated ones
(P=0.029). Furthermore, on Day 35, more aggrecan was synthesised by
cells stimulated with the combination of the growth factors TGF-β1
and IGF-1 compared to the single stimulating experiments. In
contrast to hypoxic conditions on Day 35, an accumulation of
aggrecan was detectable for TGF-β-1 stimulated cells only.
Additionally, normoxic cultivated cells showed a reduced expression
of aggrecan from Day 14 to 35 after stimulation with TGF-β1/IGF-1.
Significant differences (P=0.029) could be determined for the
stimulating experiments on both time points (Fig. 5).
Staining of aggrecan resulted in visible differences
between the culture conditions. A brighter positive staining of
TGF-β1 ± IGF-1 treated pellets was observed under hypoxia.
Furthermore, a positive staining of aggrecan was determined after
IGF-1 treatment in hypoxic cultivated pellets only (Fig. 4g–l).
Discussion
The aim of this study was to analyse the effect of
TGF-β1, IGF-1 or the combination of both growth factors on the
proliferation and synthesis rate of collagen type 1 and type 2 as
well as aggrecan of human chondrocytes cultivated as pellet
cultures under differing oxygen contents. Several studies about the
chondrogenic potential of several cell types, e.g. MSC from adipose
tissue or bone marrow, in pellet cultures were performed but only
limited studies have addressed this aspect by using chondrocytes
from human articular cartilage. In a previous study, we could show
that cells isolated from hyaline cartilage tissue express high
levels of MSC-associated markers which play an important role
during chondrogenesis (14).
Additionally, pellet cultures allow cell to cell interactions which
are similar to those in pre-cartilage condensation found during
embryonic development (16).
Based on these findings, we hypothesised that the addition of
chondrogenic growth factors could influence re-differentiation
processes of cells in a 3D environment.
After the treatment with different chondrogenic
growth factors under hypoxia and normoxia, differences in DNA
contents within the pellets could be observed. Although we used the
same initial cell number, a reduced DNA content could be observed
after 14 days under normoxia compared to hypoxic conditions. After
35 days of incubation under normoxia we could show an increase in
DNA content after treatment with TGF-β1 alone or in combination
with IGF-1. One possible explanation could be the addition of
TGF-β1 which is known to rise the proliferation of mesenchymal stem
cells during chondrogenesis (11). Nevertheless, the values of DNA
content under hypoxia could not be achieved after cultivation under
normoxic conditions. Furthermore, the single treatment with IGF-1
led to a decrease in DNA content during cultivation. We suggest
that under normoxia, differences in oxygen concentrations
predominate between cells in the periphery and in the centre of the
pellet. As a consequence, cells are exposed to an increased
oxidative stress, which results in a higher apoptosis rate. In this
context, the first results indicated an enhanced apoptosis of cells
after 14 days of incubation under normoxia which has to be examined
in a subsequent study. Additionally, normoxia resulted in an
enrichment of reactive oxygen species (ROS) which lead to a reduced
activity of especially IGF-1 (2).
In contrast, under hypoxic conditions the hypoxic-inducible
transcription factor (HIFs) proteins are permanently expressed
within chondrocytes (17) which
regulate differentiation of cells. Bohensky et al (18) showed that HIF-2α could prevent
apoptosis of chondrocytes, limiting ROS production. As our results
demonstrate, chondrocyte-pellets incubated under hypoxia showed
less evidence of cell death because of a stable DNA content.
Additionally, the chosen culture conditions of 5% oxygen is more
physiological for chondrogenic cells because in articular cartilage
the oxygen concentration is estimated to range between 1 and 5%
(17).
For the synthesis of specific ECM components,
differences in the expression between hypoxia and normoxia as well
as differences between growth factors could be observed. For the
experiments we used TGF-β1 and IGF-1 because both growth factors
play an important role for ECM formation (12,19,20). Under both culture conditions a
reduced synthesis rate of pro-collagen type 1 could be shown after
stimulation with the three growth factor compositions. For IGF-1
treated pellets a reduced synthesis rate of pro-collagen type 1
compared to TGF-β1 as well as TGF-β1/IGF-1 stimulated ones could be
shown at both time points. The difference between the combinations
was significant on Day 35. As a consequence, a more positive effect
of IGF-1 on re-differentiation of chondrocytes is observed compared
to TGF-β1.
In general, under normoxia we detected an enhanced
synthesis rate of pro-collagen type 2 and aggrecan after the
stimulation with all growth factor combinations compared to hypoxic
culture conditions. This was not unexpected because we used
chondrocytes isolated from osteoarthritic cartilage. It is well
known, that these cells are characterised by an increased
expression of collagen type 2 and other matrix components as a
response to restore ECM (21).
Additionally, after 35 days of incubation the TGF-β1
and/or IGF-1 stimulation led to an increased concentration of
pro-collagen type 2 under normoxia. These results were in
correspondence with the DNA data because proliferating chondrocytes
synthesise collagen type 2 (22).
For aggrecan, we could show an accumulation after TGF-β1 treatment
under normoxia whereas a reduced expression was determined after
TGF-β1/IGF-1 as well as IGF-1 treatment.
Under hypoxic culture conditions expression of
pro-collagen type 2 and aggrecan increased during the stimulation
with TGF-β1/IGF-1. In contrast to normoxia, a stable pro-collagen
type 2 concentration after single treatment with IGF-1 and TGF-β1
was determined. Furthermore, IGF-1 stimulation increased aggrecan
synthesis. Although IGF-1 effects on the accumulation of ECM
components were significantly reduced compared to TGF-β1/IGF-1,
IGF-1 seemed to be more efficient. Under hypoxia, expression of
collagen type 2 and aggrecan are mediated by HIF-1α which is the
survival factor in chondrocytes (23). Additionally, growth factors like
IGF-1 also influence HIF-1 activity positively (21) which could be an explanation for
superior results of aggrecan accumulation after IGF-1 treatments
under hypoxia.
Nevertheless, the IGF-1 effects on the ECM component
expression were clearly reduced under normoxia as well as under
hypoxia compared to TGF-β1 treatment. These results are in
accordance to Chubinskaya et al (9), who did not verify matrix
accumulation after single treatment of IGF-1 in alginate beads. One
reason for these effects could be the cell source used. For our
experiments, we isolated cells from osteoarthritic cartilage.
Martin et al (24)
reported an enhanced proportion of the IGF-binding protein 3 as
well as fibronectin in OA cartilage. Colocalisation of both
components in the ECM resulted in controlling of IGF-1 because of
complex formation. For this reason, the inhibitory effect of IGFBP
3 is retained (8) and IGF-1 could
not be released. This may discredit the applicability of IGFBP
pathways as targets for cartilage repair (25). Gonzalez et al (25) emphasised the impact of a
short-term TGF-β1 stimulation to inhibit IGFBP production. As a
result, applied IGF-1 could not only be stored in the ECM but also
bound on IGF-receptors to activate the intracellular pathway to
stimulate collagen type 2 and proteoglycan synthesis. Under hypoxia
an additive effect on matrix deposition after stimulation with
TGF-β1/IGF-1 was detected. We assume a positive influence of TGF-β1
on IGF-1 when using the combination of both under hypoxic culture
conditions.
In summary, treatment with IGF-1 and TGF-β1
influences the re-differentiation of chondrocytes with respect to
the accumulation of hyaline ECM compound. Nevertheless, our tests
revealed differences between cultivation conditions concerning the
oxygen content. While under normoxic conditions TGF-β1 alone leads
to a positive impact of the expression of hyaline
cartilage-specific ECM components, an additive effect of both
growth factors was found under hypoxic conditions. IGF-1 treatment
under hypoxia was more efficient than under normoxia. Thereby, our
hypothesis concerning oxygen content reduction could be confirmed
by the data of DNA concentration and ECM accumulation, i.e. under
hypoxia ROS enrichment could be limited resulting in a positive
impact for IGF-1.
Hence, a natural 3D environment in combination with
chondrogenic growth factors as well as reduced oxygen contents
could facilitate cell survival and chondrogenic re-differentiation.
However, in further studies it is recommended to analyse the
effects of growth factors in combination with normoxia or hypoxia
with chondrocytes derived from donors without osteoarthritis.
Detailed knowledge about the cell interactions in the context of
environment conditions could lead to targeted differentiation of
cartilage-derived cells into hyaline cartilage as a prerequisite
for the development of new therapeutic strategies in cartilage
repair.
Acknowledgements
The authors gratefully thank the
European Union and the Ministry of Economic Affairs, Employment and
Tourism of Mecklenburg-Vorpommern for financial support within the
research project ‘Syntero’. We thank Mrs. Frauke Winzer (Department
of Anatomy) for her technical support with immunohistochemical
preparation.
References
|
1.
|
J FarrB ColeA DhawanJ KercherS
ShermanClinical cartilage restoration: evolution and overviewClin
Orthop Relat
Res46926962705201110.1007/s11999-010-1764-z21240578
|
|
2.
|
RF LoeserAging and osteoarthritis: the
role of chondrocyte senescence and aging changes in the cartilage
matrixOsteoarthritis
Cartilage17971979200910.1016/j.joca.2009.03.00219303469
|
|
3.
|
S GiovanniniJ Diaz-RomeroT AignerP HeiniP
Mainil-VarletD NesicMicromass co-culture of human articular
chondrocytes and human bone marrow mesenchymal stem cells to
investigate stable neocartilage tissue formation in vitroEur Cell
Mater202452592010
|
|
4.
|
L PetersonHS VasiliadisM BrittbergA
LindahlAutologous chondrocyte implantation: a long-term follow-upAm
J Sports Med3811171124201010.1177/036354650935791520181804
|
|
5.
|
DG StokesG LiuR DharmavaramD HawkinsS
Piera-VelazquezSA JimenezRegulation of type-II collagen gene
expression during human chondrocyte de-differentiation and recovery
of chondrocyte-specific phenotype in culture involves Sry-type
high-mobility-group box (SOX) transcription factorsBiochem
J360461470200110.1042/0264-6021:3600461
|
|
6.
|
PD BenyaJD ShafferDedifferentiated
chondrocytes reexpress the differentiated collagen phenotype when
cultured in agarose
gelsCell30215224198210.1016/0092-8674(82)90027-77127471
|
|
7.
|
J BonaventureN KadhomL Cohen-SolalKH NgJ
BourguignonC LasselinP FreisingerReexpression of cartilage-specific
genes by dedifferentiated human articular chondrocytes cultured in
alginate beadsExp Cell
Res21297104199410.1006/excr.1994.11238174647
|
|
8.
|
PM van der KraanP BumaKT vanWB van den
BergInteraction of chondrocytes, extracellular matrix and growth
factors: relevance for articular cartilage tissue
engineeringOsteoarthritis Cartilage10631637200212479385
|
|
9.
|
S ChubinskayaA HakimiyanC PacioneA YankeL
RappoportT AignerDC RuegerRF LoeserSynergistic effect of IGF-1 and
OP-1 on matrix formation by normal and OA chondrocytes cultured in
alginate beadsOsteoarthritis
Cartilage15421430200710.1016/j.joca.2006.10.00417126570
|
|
10.
|
P GiannoniR CanceddaArticular chondrocyte
culturing for cell-based cartilage repair: needs and
perspectivesCells Tissues
Organs184115200610.1159/00009694617190975
|
|
11.
|
T FukumotoJW SperlingA SanyalJS
FitzsimmonsGG ReinholzCA ConoverSW O’DriscollCombined effects of
insulin-like growth factor-1 and transforming growth factor-beta1
on periosteal mesenchymal cells during chondrogenesis in
vitroOsteoarthritis
Cartilage115564200310.1053/joca.2002.086912505488
|
|
12.
|
PC YaegerTL MasiJL de OrtizF BinetteR
TuboJM McPhersonSynergistic action of transforming growth
factor-beta and insulin-like growth factor-I induces expression of
type II collagen and aggrecan genes in adult human articular
chondrocytesExp Cell Res237318325199710.1006/excr.1997.3781
|
|
13.
|
GJ van OschSW van der VeenP BumaHL
Verwoerd-VerhoefEffect of transforming growth factor-beta on
proteoglycan synthesis by chondrocytes in relation to
differentiation stage and the presence of pericellular matrixMatrix
Biol1741342419989840443
|
|
14.
|
A JonitzK LochnerK PetersA SalamonJ
PasoldB Mueller-HilkeD HansmannR BaderDifferentiation capacity of
human chondrocytes embedded in alginate matrixConnect Tissue
Res52503511201110.3109/03008207.2011.59367321787134
|
|
15.
|
PA GuerneF BlancoA KaelinA DesgeorgesM
LotzGrowth factor responsiveness of human articular chondrocytes in
aging and developmentArthritis
Rheum38960968199510.1002/art.17803807127612045
|
|
16.
|
L ZhangP SuC XuJ YangW YuD
HuangChondrogenic differentiation of human mesenchymal stem cells:
a comparison between micromass and pellet culture systemsBiotechnol
Lett3213391346201010.1007/s10529-010-0293-x20464452
|
|
17.
|
JE LafontLack of oxygen in articular
cartilage: consequences for chondrocyte biologyInt J Exp
Pathol9199106201010.1111/j.1365-2613.2010.00707.x20384821
|
|
18.
|
J BohenskySP TerkhornTA FreemanCS AdamsJA
GarciaIM ShapiroV SrinivasRegulation of autophagy in human and
murine cartilage: hypoxia-inducible factor 2 suppresses chondrocyte
autophagyArthritis Rheum6014061415200910.1002/art.2444419404942
|
|
19.
|
JA TylerInsulin-like growth factor 1 can
decrease degradation and promote synthesis of proteoglycan in
cartilage exposed to cytokinesBiochem J26054354819892788408
|
|
20.
|
TI MoralesThe insulin-like growth factor
binding proteins in uncultured human cartilage: increases in
insulin-like growth factor binding protein 3 during
osteoarthritisArthritis Rheum4623582367200210.1002/art.10482
|
|
21.
|
D PfanderK GelseHypoxia and
osteoarthritis: how chondrocytes survive hypoxic environmentsCurr
Opin Rheumatol19457462200710.1097/BOR.0b013e3282ba569317762611
|
|
22.
|
E SchipaniPosttranslational modifications
of collagens as targets of hypoxia and Hif-1alpha in endochondral
bone developmentAnn NY Acad
Sci1192317321201010.1111/j.1749-6632.2009.05236.x20392253
|
|
23.
|
E DuvalS LeclercqJM ElissaldeM DemoorP
GaleraK BoumedieneHypoxia-inducible factor 1alpha inhibits the
fibroblast-like markers type I and type II collagen during
hypoxia-induced chondrocyte redifferentiation: hypoxia not only
induces type II collagen and aggrecan, but it also inhibits type I
and type II collagen in the hypoxia-inducible factor
1alpha-dependent redifferentiation of chondrocytesArthritis
Rheum60303830482009
|
|
24.
|
JA MartinBA MillerMB ScherbLA LembkeJA
BuckwalterCo-localization of insulin-like growth factor binding
protein 3 and fibronectin in human articular
cartilageOsteoarthritis
Cartilage10556563200210.1053/joca.2002.079112127836
|
|
25.
|
C GonzalezKG Auw YangJH SchwabJS
FitzsimmonsMM ReinholzZT ReschLK BaleVR ClemensCA ConoverSW
O’DriscollGG ReinholzTransforming growth factor-beta1 modulates
insulin-like growth factor binding protein-4 expression and
proteolysis in cultured periosteal explantsGrowth Horm IGF
Res208186201010.1016/j.ghir.2009.06.00219656700
|