Introduction
A number of natural factors and nutritional
supplements have long been believed to promote skeletal health.
However, it is only relatively recently that these effects have
been ratified in animal models and in vitro systems and in a
few instances such as vitamin K2, in clinical trials
(1). However, their mechanisms of
action remain poorly defined. We have previously investigated the
mode of action of a number of micronutrients and nutritional
factors on bone cells including quercetin, zinc, vitamin
K2, honokiol and the carotenoid β-cryptoxanthin
(2–7). Interestingly, we reported that all
of these agents appear to utilize a common centralized mechanism
for rebalancing bone turnover by differentially regulating bone
formation and resorption through suppression of the nuclear
factor-κB (NF-κB) signal transduction pathway (2–7).
Indeed NF-κB is established as a critical component
of osteoclast differentiation and activity and is an essential
signal mediated through association of receptor activator of NF-κB
(RANKL) with its receptor RANK (8,9).
NF-κB antagonists have been demonstrated to ameliorate bone loss
associated with estrogen deficiency in mice, a model of
postmenopausal osteoporosis (10), in animal models of rheumatoid
arthritis (11) and in bone loss
associated with multiple myeloma (12).
More recently the role of NF-κB signaling in
osteoblast differentiation has been investigated and found to be
potently inhibitory. This finding was consistent with the
well-established inhibitory action on osteoblastic differentiation
and mineralization of the inflammatory cytokine tumor necrosis
factor-α (TNF-α), a potent activator of NF-κB (13). In fact, we have demonstrated in
vivo that even physiological concentrations of TNF-α achieve a
magnitude sufficient to significantly impede bone formation and
reduce maximum achievable peak bone mineral density (BMD) in mice
in vivo (14). Other
studies have reported that TNF-α suppresses bone formation in
models of fracture repair (15,16) while conditional inactivation of
NF-κB in osteoblasts promotes bone formation and ameliorates
ovariectomy-induced bone loss (17). TNF-α, through NF-κB activation
impedes the transforming growth factor-β (TGF-β)-induced commitment
of early mesenchymal stem cells along the osteoblast pathway as
well as suppressing BMP-induced bone formation and osteoblast
differentiation, by intersecting with and antagonizing the
activation of Smad-signal transduction (2–7,14,18). Smad activity in the osteoblast is
in turn regulated by Smurf1-induced proteasomal degradation
(19) as well as by upregulation
of the Smad activation antagonist Smad7 (20).
p-Hydroxycinnamic acid (HCA) is a carotene
that is derived from cinnamic acid found in plants and fruits. HCA
has been found to stimulate bone formation in rat femoral tissues
in organ culture ex vivo (21) and to inhibit the differentiation
of primary bone marrow cells into osteoclasts in vitro
(22) suggesting dual anabolic
and anti-catabolic activities. Furthermore, HCA has been used to
effectively ameliorate ovariectomy-induced bone loss in rats
(23) as well as bone loss
associated with streptozotocin-induced diabetes in rats (24).
In this study, we examined the mechanism pertaining
to the bone anabolic and anticatabolic activity of HCA on bone
cells. Our data reveal that like other agents endowed with dual
anabolic and anticatabolic activities, HCA functions as a natural
NF-κB antagonist, suppressing RANKL-induced NF-κB signaling in
osteoclast precursors and relieving the inhibitory actions of TNF-α
on the pro-anabolic Smad pathway.
Materials and methods
Materials
α-minimal essential medium (α-MEM) and antibiotics
(penicillin and streptomycin) were purchased from Invitrogen Corp.
(Carlsbad, CA). Fetal bovine serum (FBS) was from Hyclone. RANKL,
TGF-β, TNF-α and BMP-2 were from R&D Systems (Minneapolis, MN).
HCA, mouse anti-poly-histidine antibody and all other reagents were
purchased from the Sigma Chemical Corporation (St. Louis, MO)
unless otherwise specified.
Cell culture
The preosteoblastic cell line MC3T3-E1, clone 14
(MC3T3) and the monocytic cell line RAW264.7 were purchased from
the American Type Culture Collection (Manassas, VA) and cultured as
described previously (7,14).
Osteoblast differentiation assays and
Alizarin Red-S staining
MC3T3 cells or primary bone marrow stromal cells
were plated and cultured for 72 h in α-MEM (1.0 ml/well) containing
10% FBS in 12-well dishes at a density of 1.0x105
cells/well. The medium was aspirated and changed to mineralization
medium (α-MEM supplemented with 10% FBS, L-ascorbic acid (100
µg/ml) and 4 mM β-glycerophosphate) as previously described
(14,25). Vehicle or HCA was added at a dose
of 0.01 to 10 µM and cells were replenished with fresh medium every
3 days. At 17–18 days cells were rinsed with PBS and calcium
deposition was visualized by fixing the cells in 75% ethanol for 30
min at 4˚C followed by staining with Alizarin Red-S (40 mM, pH 6.2)
for 30 min at room temperature. Excess stain was removed by copious
washing with distilled water. Plates were imaged using a flatbed
scanner (Epson Perfection 1660 Photo), and quantitated using ImageJ
(26). To quantify mineralization
Alizarin Red-S was eluted from cultures by incubation in 10%
cetylpyridinium chloride solution. After complete elution,
absorbance was read at 570 nm on a microtiter plate reader.
Osteoclastogenesis assays and TRAP
staining
RAW264.7 cells were cultured in 96-well plates in
α-MEM supplemented with 10% FBS, 100 IU/ml penicillin, and 100
μg/ml streptomycin at a density of 1x104 cells/well.
Cells were cultured for 6 days with RANKL (30 ng/ml) pre-incubated
for 10 min with crosslinking anti-polyhistidine antibody (2.5
μg/ml) to induce osteoclast formation. HCA was added in the range
of 0.1–100 μM. After 6 days of culture, the cells were fixed and
stained for tartrate resistant acid phosphatase (TRAP) activity
using a leukocyte acid phosphatase kit (Sigma). TRAP+
cells with three or more nuclei were defined as osteoclasts and
were quantitated under light microscopy and 5 wells/group
averaged.
NF-κB and Smad reporter constructs and
luciferase assays
The NF-κB responsive reporter pNF-κB-Luc (BD
Biosciences) and the Smad responsive reporter pGL3-Smad were used
as previously described (14).
The Smad reporter is responsive to both TGF-β- and to BMP-induced
Smad species. Briefly, reporter plasmids were transfected into
MC3T3 or RAW264.7 cells (1x105 cells/well) using
Lipofectamine 2000 reagent (Invitrogen) in α-MEM without FBS and
antibiotics. Five hours later the medium was changed to α-MEM
containing 10% FBS plus antibiotics and the cells were treated with
TNF-α (MC3T3) or RANKL (RAW264.7) to stimulate NF-κB activity, or
with TGF-β (1 ng/ml) or BMP-2 (0.5 µg/ml) to stimulate Smad
activity and treated with or without TNF-α. Parallel groups
received vehicle or HCA in the dose range of 0.1–100 μM. Cells were
extracted with passive lysis buffer (Promega, Madison, WI) 24 h
later, and luciferase activity was measured using the Luciferase
Assay System of Promega, on a microplate luminometer (Turner
Designs, Sunnyvale, CA).
Statistical analysis
Statistical significance was determined using the
GraphPad InStat version 3 for Windows XP (GraphPad Software, Inc.,
La Jolla, CA). Multiple comparisons were performed by one-way
analysis of variance (ANOVA) with the Tukey-Kramer multiple
comparisons post-test. P<0.05 was considered statistically
significant.
Results
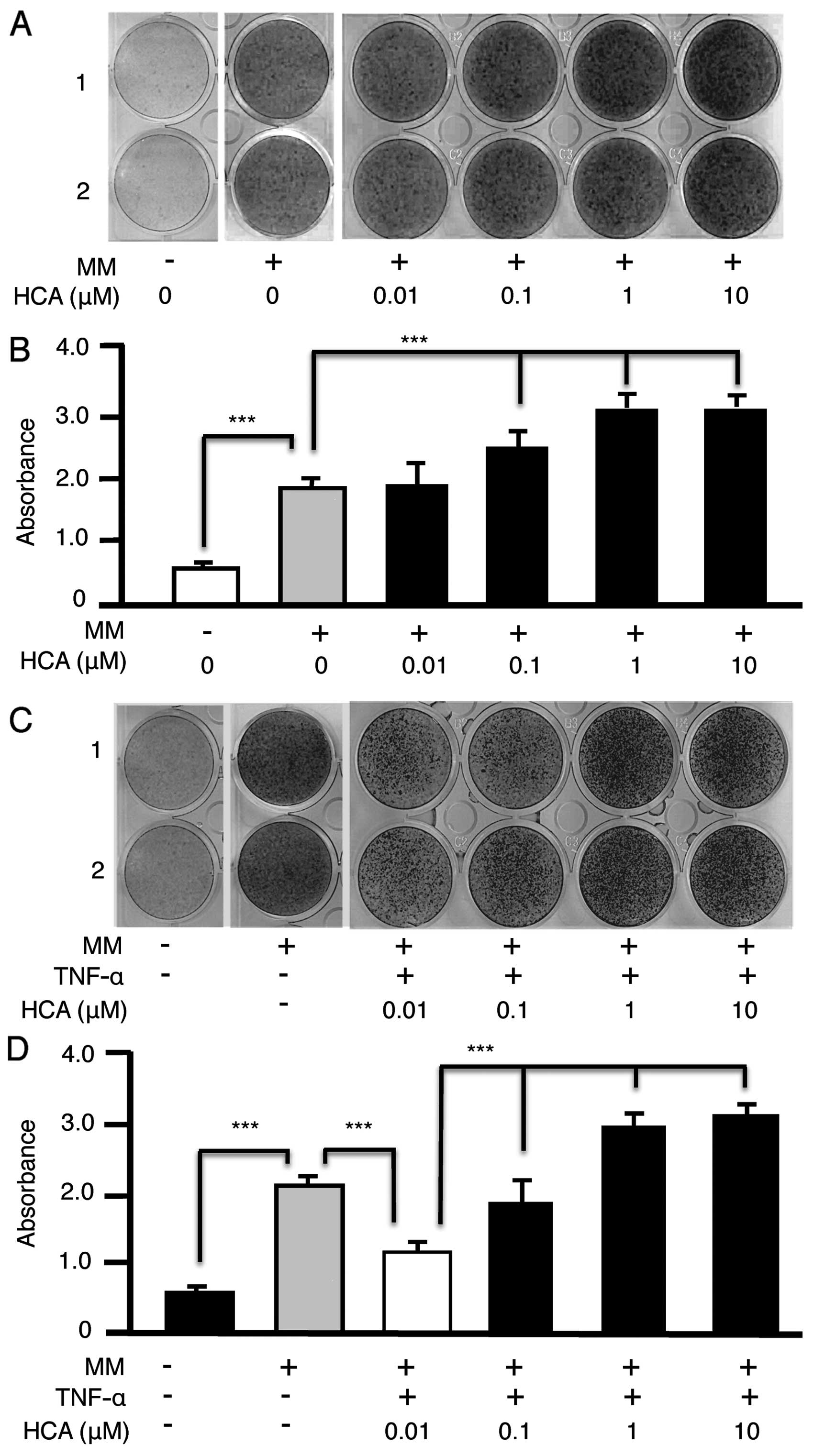
HCA augments the differentiation of MC3T3
preosteoblasts into mineralizing osteoblasts
HCA has been demonstrated to promote differentiation
of primary osteoblasts in vitro. To validate the MC3T3
preosteoblastic cell line model for mechanistic investigations of
HCA action we treated MC3T3 cells with HCA (0.01–10 μM) in
mineralizing medium (MM) for 21 days. Cultures were stained with
Alizarin Red-S to visualize calcium deposition (Fig. 1A). Alizarin Red-S was subsequently
eluted from the wells and quantified by spectrophotometry (Fig. 1B). HCA dose-dependently increased
the mineralization of MC3T3 cells ratifying their use for further
detailed mechanistic studies.
HCA alleviates the inhibitory effect of
TNF-α on MC3T3 mineralization
Because TNF-α-induced NF-κB signaling potently
inhibits osteoblastic differentiation in vivo and in
vitro (13,14) we investigated whether HCA could
relieve the inhibitory effect of TNF-α on MC3T3 mineralization. HCA
dose dependently reversed the inhibitory effect of TNF-α visualized
by Alizarin Red-S staining (Fig.
1C) and quantified by spectrophotometry (Fig. 1D).
HCA has no direct effect on basal or
cytokine-induced Smad activity but antagonizes the inhibitory
effect of TNF-α on Smad activity
We next investigated whether HCA regulates the bone
anabolic Smad signaling pathway in preosteoblasts, either directly
by indirectly. To achieve this we transfected MC3T3 cells with a
Smad responsive luciferase reporter. The Smad agonists TGF-β
(Fig. 2A) and BMP-2 (Fig. 2B) upregulated Smad reporter
activity, however HCA had no direct additive effects on baseline or
cytokine-induced Smad induction. However, when Smad activation was
potently repressed by addition of TNF-α, HCA significantly and
dose-dependently (0.1–100 µM) reversed the inhibitory action of
TNF-α (Fig. 2C). These data
suggest an indirect stimulatory effect of HCA by relieving
TNF-α-induced inhibitory effects on Smad signal transduction.
HCA suppresses TNF-α-induced NF-κB
activation in MC3T3 cells
Because NF-κB-signal transduction is potently
inhibitory to anabolic signals such as Smad (2–7,14,18) and TNF-α is a potent NF-κB
activator we further investigated whether HCA suppresses
TNF-α-induced NF-κB activation in MC3T3 pre-osteoblasts. MC3T3
cells were transiently transfected with an NF-κB luciferase
reporter and basal or cytokine (TNF-α) stimulated NF-κB activity
quantified in the presence or absence of HCA. While HCA failed to
modulate basal NF-κB activity it dose-dependently (0.1–100 µM)
downregulated TNF-α-induced NF-κB reporter activity (Fig. 3).
These data suggest that HCA promotes osteoblast
differentiation in vivo by alleviating the inhibitory action
of endogenous NF-κB-inducing antagonists such as TNF-α on
pro-osteoblastic pathways, such as Smad.
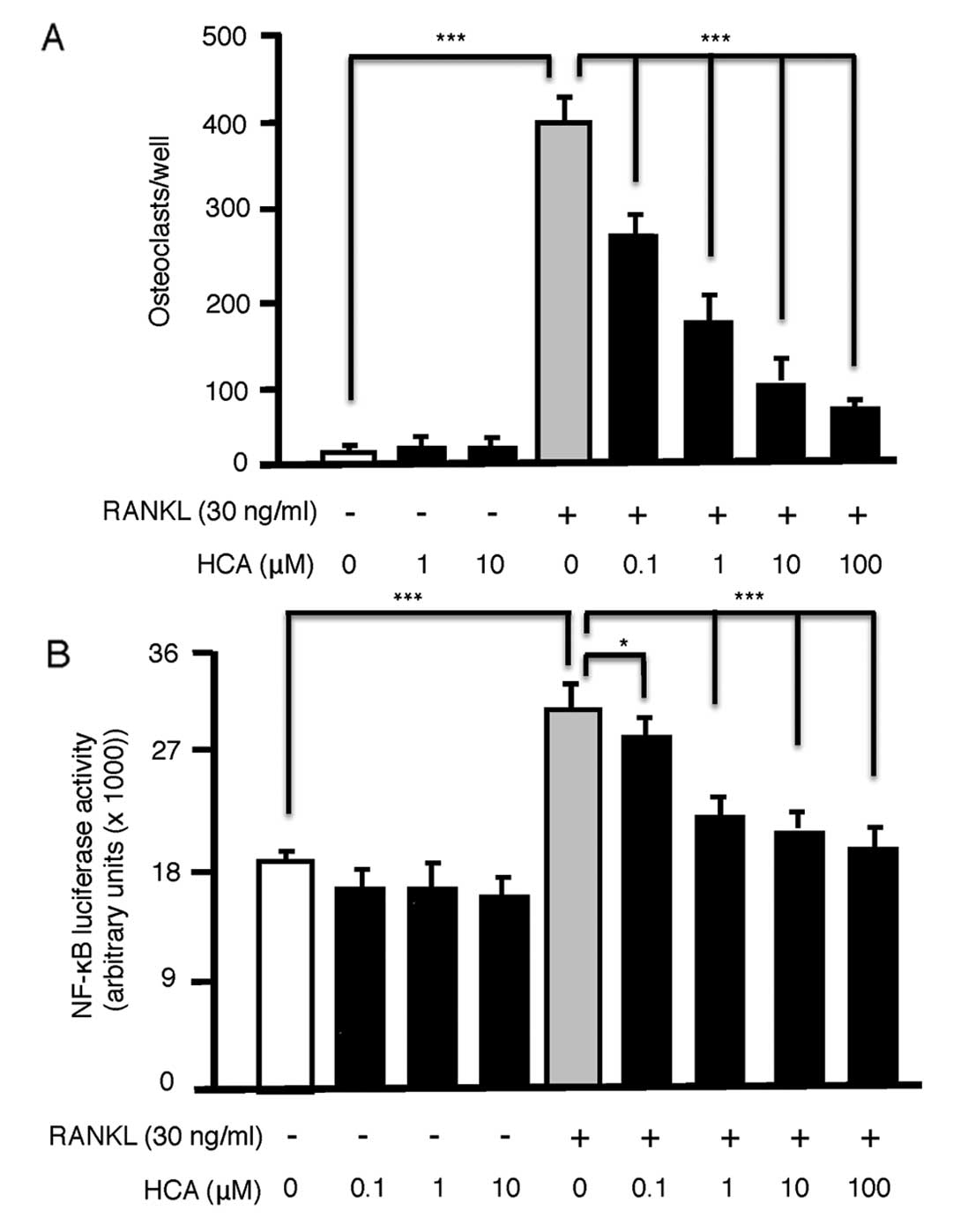
HCA dose-dependently inhibits osteoclast
formation in RAW264.7 cells by antagonizing NF-κB activation
Although HCA is known to promote osteoblastic bone
formation, its action on osteoclastic bone resorption is reported
to be repressive. To further examine the mechanism of HCA action on
osteoclasts we induced the RAW264.7 osteoclast precursor cell line
to differentiate into osteoclasts by exposure to the key
osteoclastogenic cytokine RANKL (30 ng/ml) for 7 days. RANKL
elicited significant osteoclast formation, which was significantly
and dose-dependently (0.1–100 μM) inhibited by addition of HCA
(Fig. 4A). HCA had no observable
toxic effect on RAW264.7 cell proliferation in the absence of
RANKL, as precursors continued to divide over 7 days filling the
wells with TRAP-negative mononucleated cells. These data suggest a
specific inhibitory effect of HCA on osteoclast differentiation by
RANKL.
As a primary signal mediated via RANKL and required
for osteoclast differentiation is NF-κB, a pathway established in
the studies above to be antagonized by HCA in osteoblasts, we
further examined the effect of HCA on NF-κB activation by RANKL in
RAW264.7 cells. HCA had no effect on basal NF-κB activation but
dose-dependently (0.1–100 μM) inhibited RANKL-induced NF-κB
activation (Fig. 4B).
Taken together our data suggest that HCA promotes
osteoblast differentiation and/or mineralizing activity and
suppresses osteoclast differentiation. These events are likely
mediated though suppression of NF-κB activation.
Discussion
How HCA induces bone-anabolic activity and inhibits
osteoclastic bone loss is unclear. Our data suggest that HCA
functions as a NF-κB antagonist, suppressing RANKL-induced NF-κB
signaling in osteoclast precursors while relieving the inhibitory
actions of TNF-α on BMP- and TGF-β-induced Smad activation, signals
that mediate anabolic effects in osteoblasts.
Although TNF-α was added exogenously in our in
vitro culture systems as a proof of concept, we have previously
demonstrated that in mice in vivo, the concentrations of
basal TNF-α are of a sufficient magnitude to suppress basal bone
formation. These concentrations however, did not achieve a level
capable of augmenting RANKL-induced osteoclastic bone resorption
(14). By contrast, under
inflammatory conditions characteristic of rheumatoid arthritis and
estrogen deficiency, TNF-α achieves concentrations that are able to
stimulate RANKL-induced osteoclast formation as well as suppress
osteoblastic bone formation. In fact, ovariectomy-induced bone loss
is a consequence of an imbalance between osteoclastic bone
resorption, which is significantly elevated and osteoblastic bone
formation, that although also elevated does not reach a magnitude
capable of compensating for the high rates of bone resorption
(27). TNF-α may play a central
role in both processes. On the osteoclastic side, TNF-α is reported
to amplify the activity of RANKL in osteoclast precursors (28,29) by synergizing at the level of
signal transduction (30). TNF-α
further activates the resorptive activity of osteoclasts
independently and synergistically with RANKL (31). At the other end of the spectrum
the compensatory rise in bone formation necessary to maintain
homeostasis between bone formation and resorption is impeded by
inhibitory cytokines, including TNF-α. Our data suggest that HCA
may thus prevent the ovariectomy-induced bone loss, as previously
reported (23), by not only
reducing TNF-α- and RANKL-induced osteoclastic bone resorption, but
also by relieving the suppressive effect of TNF-α on bone
formation.
In conclusion, our study supports an important
protective action of HCA on the skeleton by promoting bone
formation under basal conditions and by both suppressing bone
resorption and promoting bone formation in inflammatory
contexts.
References
|
1.
|
MK SheaSL BoothUpdate on the role of
vitamin K in skeletal healthNutr
Rev66549557200810.1111/j.1753-4887.2008.00106.x18826451
|
|
2.
|
M YamaguchiMN WeitzmannQuercetin, a potent
suppressor of NF-κB and Smad activation in osteoblastsInt J Mol
Med28521525201121769418
|
|
3.
|
M YamaguchiMN WeitzmannZinc stimulates
osteoblastogenesis and suppresses osteoclastogenesis by
antagonizing NF-kappaB activationMol Cell
Biochem355179186201110.1007/s11010-011-0852-z21533765
|
|
4.
|
M YamaguchiMN WeitzmannVitamin
K2 stimulates osteoblastogenesis and suppresses
osteoclastogenesis by suppressing NF-κB activationInt J Mol
Med273142011
|
|
5.
|
M YamaguchiJL ArbiserMN WeitzmannHonokiol
stimulates osteoblastogenesis by suppressing NF-κB activationInt J
Mol Med2810491053201121887456
|
|
6.
|
M YamaguchiMN WeitzmannThe bone anabolic
carotenoids p-hydroxycinnamic acid and β-cryptoxanthin
antagonize NF-κB activation in MC3T3 preosteoblastsMol Med
Rep26416442009
|
|
7.
|
M YamaguchiMN WeitzmannThe bone anabolic
carotenoid β-cryptoxanthin enhances transforming growth
factor-β1-induced Smad activation in MC3T3 preosteoblastsInt J Mol
Med246716752009
|
|
8.
|
S VairaM AlhawagriI AnwisyeH KitauraR
FaccioDV NovackRelA/p65 promotes osteoclast differentiation by
blocking a RANKL-induced apoptotic JNK pathway in miceJ Clin
Invest11820882097200818464930
|
|
9.
|
BF BoyceL XingG FranzosoU
SiebenlistRequired and nonessential functions of nuclear
factor-kappaB in bone
cellsBone25137139199910.1016/S8756-3282(99)00105-210423039
|
|
10.
|
K StraitY LiDL DillehayMN
WeitzmannSuppression of NF-κB activation blocks osteoclastic bone
resorption during estrogen deficiencyInt J Mol Med215215252008
|
|
11.
|
S DaiT HirayamaS AbbasY Abu-AmerThe
IkappaB kinase (IKK) inhibitor, NEMO-binding domain peptide, blocks
osteoclastogenesis and bone erosion in inflammatory arthritisJ Biol
Chem2793721937222200410.1074/jbc.C40025820015252035
|
|
12.
|
R FengG AndersonG XiaoSDX-308, a
nonsteroidal anti-inflammatory agent, inhibits NF-kappaB activity,
resulting in strong inhibition of osteoclast formation/activity and
multiple myeloma cell
growthBlood10921302138200710.1182/blood-2006-07-02745817095620
|
|
13.
|
MS NanesTumor necrosis factor-alpha:
molecular and cellular mechanisms in skeletal
pathologyGene321115200310.1016/S0378-1119(03)00841-214636987
|
|
14.
|
Y LiA LiK StraitH ZhangMS NanesMN
WeitzmannEndogenous TNFalpha lowers maximum peak bone mass and
inhibits osteoblastic Smad activation, through NF-kappaBJ Bone
Miner Res22646655200710.1359/jbmr.07012117266397
|
|
15.
|
EC WahlJ AronsonL LiuRestoration of
regenerative osteoblastogenesis in aged mice: modulation of TNFJ
Bone Miner Res25114123201010.1359/jbmr.09070819580462
|
|
16.
|
EC WahlJ AronsonL LiuDirect bone formation
during distraction osteogenesis does not require TNFalpha receptors
and elevated serum TNFalpha fails to inhibit bone formation in
TNFR1 deficient miceBone46410417201010.1016/j.bone.2009.09.011
|
|
17.
|
J ChangZ WangE TangInhibition of
osteoblastic bone formation by nuclear factor-kappaBNat
Med15682689200910.1038/nm.195419448637
|
|
18.
|
M YamazakiH FukushimaM ShinTumor necrosis
factor alpha represses bone morphogenetic protein (BMP) signaling
by interfering with the DNA binding of Smads through the activation
of NF-kappaBJ Biol
Chem2843598735995200910.1074/jbc.M109.07054019854828
|
|
19.
|
R GuoM YamashitaQ ZhangUbiquitin ligase
Smurf1 mediates tumor necrosis factor-induced systemic bone loss by
promoting proteasomal degradation of bone morphogenetic signaling
proteinsJ Biol
Chem2832308423092200810.1074/jbc.M70984820018567580
|
|
20.
|
RA EliseevEM SchwarzMJ ZuscikRJ O’KeefeH
DrissiRN RosierSmad7 mediates inhibition of Saos2 osteosarcoma cell
differentiation by NFkappaBExp Cell
Res3124050200610.1016/j.yexcr.2005.09.01616259979
|
|
21.
|
YL LaiM YamaguchiPhytocomponent
p-hydroxycinnamic acid stimulates bone formation and
inhibits bone resorption in rat femoral tissues in vitroMol Cell
Biochem2924552200617036165
|
|
22.
|
YL LaiM YamaguchiPhytocomponent
p-hydroxycinnamic acid inhibits osteoclast-like cell
formation in mouse bone marrow culturesInt J Mol
Med191231282007
|
|
23.
|
M YamaguchiYL LaiS UchiyamaT NakagawaOral
administration of phytocomponent p-hydroxycinnamic acid
prevents bone loss in ovariectomized ratsMol Cell
Biochem31131362008
|
|
24.
|
M YamaguchiS UchiyamaYL LaiOral
administration of phytocomponent p-hydroxycinnamic acid has
a preventive effect on bone loss in streptozotocin-induced diabetic
ratsInt J Mol Med198038072007
|
|
25.
|
E SugimotoM YamaguchiAnabolic effect of
genistein in osteoblastic MC3T3-E1 cellsInt J Mol
Med5515520200010762655
|
|
26.
|
MD AbramoffPJ MagelhaesSJ RamImage
processing with ImageJBiophotonics Int1136422004
|
|
27.
|
MN WeitzmannR PacificiEstrogen deficiency
and bone loss: an inflammatory taleJ Clin
Invest11611861194200610.1172/JCI2855016670759
|
|
28.
|
S CenciMN WeitzmannC RoggiaEstrogen
deficiency induces bone loss by enhancing T-cell production of
TNF-alphaJ Clin Invest10612291237200010.1172/JCI1106611086024
|
|
29.
|
J LamS TakeshitaJE BarkerO KanagawaFP
RossSL TeitelbaumTNF-alpha induces osteoclastogenesis by direct
stimulation of macrophages exposed to permissive levels of RANK
ligandJ Clin Invest10614811488200010.1172/JCI1117611120755
|
|
30.
|
YH ZhangA HeulsmannMM TondraviA MukherjeeY
Abu-AmerTumor necrosis factor-alpha (TNF) stimulates RANKL-induced
osteoclastogenesis via coupling of TNF type 1 receptor and RANK
signaling pathwaysJ Biol
Chem276563568200110.1074/jbc.M008198200
|
|
31.
|
K FullerC MurphyB KirsteinSW FoxTJ
ChambersTNFalpha potently activates osteoclasts, through a direct
action independent of and strongly synergistic with
RANKLEndocrinology14311081118200211861538
|