Introduction
Neurofibromatosis type 1 (NF-1) is a common genetic
disorder that is transmitted by autosomal dominant inheritance, and
is diagnosed when two of the following signs are found in
individuals: six or more cafe-au-lait spots over 5 mm in greatest
diameter during prepubertal period, freckling in the axillary or
inguinal region 2–3 mm in diameter, two or more Lisch nodules in
the iris, optic gliomas, two or more neurofibromas or one plexiform
neurofibroma, skeletal abnormalities such as a distinctive osseous
lesion like sphenoid dysplasia or pseudo-arthritis, or family
history of a first degree relative with NF-1 (1).
Short stature, defined as a height that is less than
2 standard deviations to the population mean, has been known to be
one of the most common complications in NF-1. Although it is known
to be associated with several clinical risk factors related to
NF-1, the exact mechanism for short stature is not yet defined
(2).
Growth hormone insensitivity syndrome (GHIS) is
known to be a very rare genetic disorder with an autosomal
recessive inheritance, and is characterized by normal or elevated
GH concentration, very low insulin-like growth factor 1 (IGF-1) and
IGF-binding protein 3 (IGFBP3) levels in serum. GH binds to its
receptor and then activates an intracellular signal transduction
pathway, leading to the production of IGF-1. IGF-1 mediates the
actions of GH and performs a negative feedback of GH secretion at
the pituitary level. Any defect in this pathway results in GHIS.
Consequently, patients with this syndrome have postnatal growth
failure, leading to short adult stature, and abnormal facial
morphologies such as frontal bossing of the forehead, hypoplasia of
the midfacies and depressed nasal bridge (3,4).
GHIS was first reported by Z. Laron in two siblings
from an Oriental Jewish family, since then many cases have been
described and most of them revealed gene defects of various
proteins involved in the GH - IGF-1 signaling cascade (5,6).
We examined a patient with GHIS accompanied by NF-1,
the first reported case with biochemical and molecular
analysis.
Materials and methods
Patient
A pediatric patient with growth retardation was
brought to the department of pediatric endocrinology in our
hospital. The patient’s birth, medical, nutritional and family
history were reviewed. His height, weight and head circumference
were measured and assessed by standard deviation score (SDS) as
well as the assessment of growth velocity since birth. Informed
consent was obtained from three family members who participated in
the study.
Biochemical and imaging study
GH stimulation was performed. The basal serum
concentrations of GH, IGF-1, IGFBP3, thyroid stimulating hormone
(TSH) with free T4, luteinizing hormone (LH), follicle stimulating
hormone (FSH) and cortisol were measured. Serum concentrations of
GH and cortisol were measured every 30 min during 120 min after
stimulation of insulin (0.1 IU/kg, intravenously). Data of serum GH
concentrations was also obtained from age-matched control group
(n=10) and compared to those of the patient. This test was planned
to be discontinued if hypoglycemic symptoms occurred or when serum
glucose level after stimulation with insulin was 45 mg/dl or
<50% of basal level. Brain magnetic resonance imaging (MRI)
scanning was also performed in order to investigate abnormal brain
lesions associated with growth retardation.
Extraction of genomic DNA and
total-RNA
Genomic DNA (gDNA) was isolated from the patient and
his family members using Accuprep Genomic DNA extraction kit
(Bioneer, Seoul, Korea), according to the manufacturer’s
instructions. Total-RNA extraction was carried out in two steps;
leukocytes from whole blood were obtained in step I, RNA from the
leukocytes was extracted in step II subsequently. Leukocytes were
isolated from the patient, his family members and healthy control
using Ficoll-Paque Plus (GE Healthcare, USA), according to the
manufacturer’s instructions.
Reverse-transcription polymerase chain
reaction (RT-PCR)
cDNA was synthesized from 4 μg of total-RNA
in a 20 μl reaction mixture containing ImProm-II™ Reverse
Transcription System (Promega, Madison, WI, USA). First strand cDNA
was amplified using PCR. This mixture was placed at 42°C for 60
min. The synthesized cDNA was incubated at 70°C for 10 min, and
then stored at −80°C. To verify the amplification, the PCR products
were subsequently examined by 1.2% agarose gel electrophoresis.
Direct DNA sequencing
PCR amplification of all growth hormone receptor
(GHR) exons, including the flanking intron regions, was performed
on extracted DNA and RNA using previously described amplification
primers and cycling conditions, as described by Vidal et al
(7) with some modifications.
Primer sequences used for amplification of human GHR gene
fragments are listed in Table
I.
 | Table I.Oligonucleotide sequences of the
GHR primer sets used in this study. |
Table I.
Oligonucleotide sequences of the
GHR primer sets used in this study.
| Sense primer
(5′→3′) | | Antisense primer
(5′→3′) | Amplified
region |
|---|
| Exon 2 F |
GTCTGCTTTTAATTGCTGGGC | Exon 2 R |
ACACTGAGGGTGGAAATGGA | Exon 2 |
| Exon 3 F |
CCTCTTTCTGTTTCAGCCAC | Exon 3 R |
GGATAGTAGCTTAATTACACTA | Exon 3 |
| Exon 4 F |
AGGATCACATATGACTCACCT | Exon 4 R |
AGTGTACTTTAGTAGGTACATC | Exon 4 |
| Exon 5 F |
TAAGCTACAACATGATTTTTGG | Exon 5 R |
TTAGTCTAAAACTATGTCAAAG | Exon 5 |
| Exon 6 F |
GTGTCTGTCTGTGTACTAATG | Exon 6 R |
AGAAAAGTCAAAGTGTAAGGTG | Exon 6 |
| Exon 7 F |
TAGTGTTCATTGGCATTGAG | Exon 7 R |
ACAAAAGCCAGGTTAGCTAC | Exon 7 |
| Exon 8 F |
AAACTGTGCTTCAACTAGTCG | Exon 8 R |
GGTCTAACACAACTGGTACA | Exon 8 |
| Exon 9 F |
GAATATGTAGCTTTTAAGATGTC | Exon 9 R |
CATATGACAGGAGTCTTCAGGTG | Exon 9 |
| Exon 10 F |
GAGTTTCTTTTCATAGATCTTC | Exon 10 R |
GGTTTAAACATTGTTTTGGC | Exon 10 |
| Exon 10-1 F |
GATCTTCATTTTCTTTCTAT | Exon 10-1 R |
CTACCTGCTGGTGTAATGTC | Exon 10-1 |
| Exon 10-2 F |
CATCGACTTTTATGCCCAGG | Exon 10-2 R |
ATGAATGGAGGTATAGTCTGG | Exon 10-2 |
| Exon 10-3 F |
CATGTTCCAGGTTCTGAGAT | Exon 10-3 R |
GGTTTAAACATTGTTTTGGC | Exon 10-3 |
The PCR amplifications were performed and the
products were purified with a QIA-quick-PCR-purification kit
(Qiagen, Germantown, MD, USA), and then directly sequenced with an
ABI-3700 automated DNA sequencer (Applied-Biosystems, Foster City,
CA, USA).
Results
Patient
The patient was a 2-year-old boy, who was born by
normal vaginal spontaneous delivery with normal birth length and
weight. No abnormal findings were observed on the neonatal
screening test. The patient’s mother and maternal grandfather were
diagnosed with NF-1 (Fig. 1).
Also, the patient was diagnosed with NF-1 at the age of 6 months
when the patient had 6 or more cafe-au-lait spots with >5 mm in
greatest diameter on the body. The patient showed normal hair,
frontal bossing and a depressed nasal bridge. On admission, 75 cm
of height (<3 percentile), 8.3 kg of weight (<3 percentile),
and 48 cm of head circumference (25–50 percentile) were recorded.
Since birth, the patient had showed reduced growth velocity,
resulting in postnatal growth failure (Fig. 2). The heights of the parents and
maternal grandfather were within normal range.
Biochemical and imaging study
The GH stimulation showed that GH concentration was
4.13 ng/ml at 0 min but 94 ng/ml at 60 min. The rapid increase in
GH serum level following insulin stimulation revealed the
significant difference from the general pattern of normal control
(Fig. 3). The patient’s serum
IGF-1 and IGFBP-3 levels were low, <25 ng/ml and <1.2 mg/l,
respectively. On the brain MRI scanning, there was no evidence of
other abnormalities including brain tumor.
Molecular studies
No germline mutation of the GHR gene
including intron-exon boundaries was found in the patient and his
family members. A complete deletion of exon 7 (c.619_784 del) of
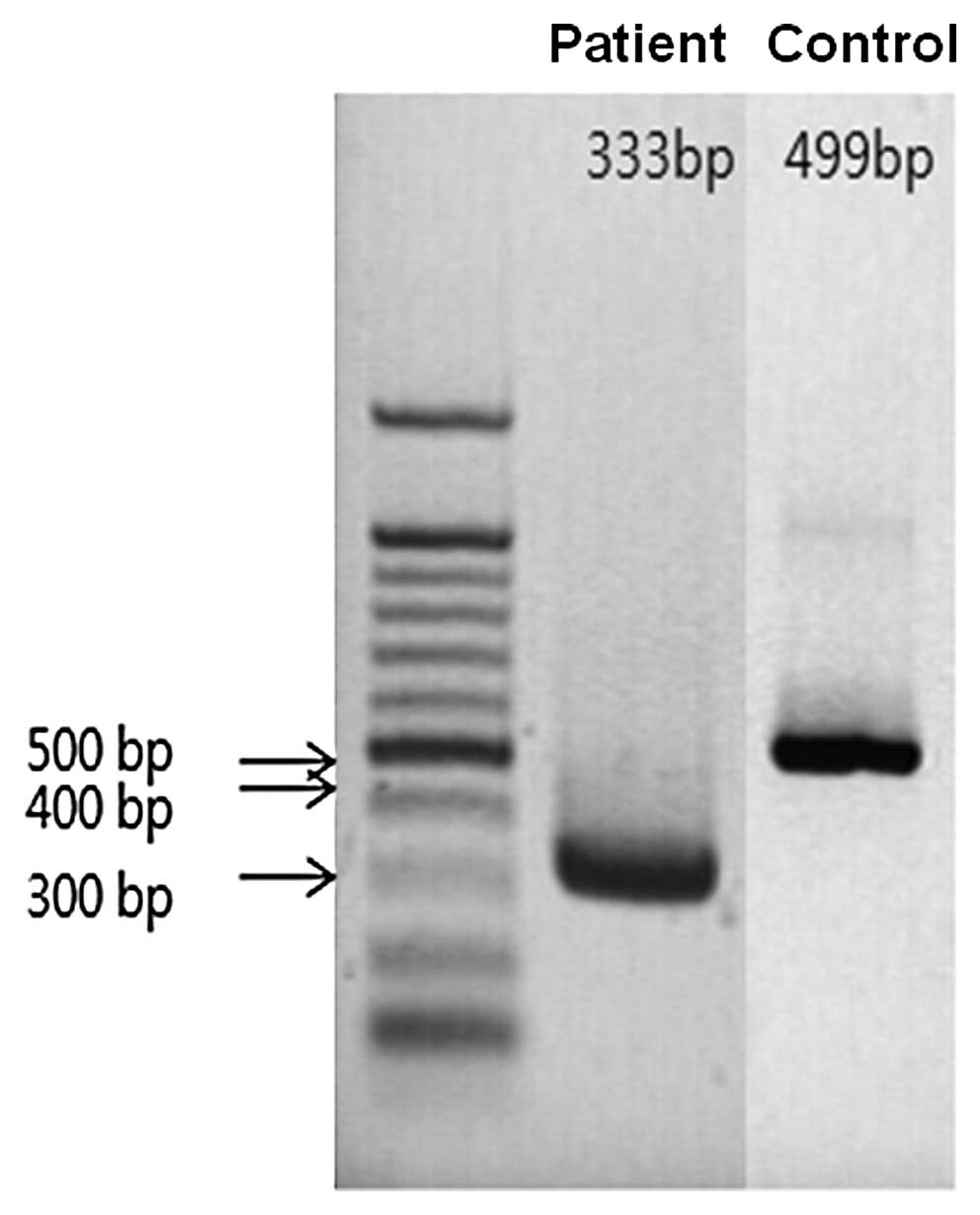
the GHR mRNA was observed. By analysis of agarose gel
electrophoresis, a deletion of 166 base pairs in exon 7 was found
(Fig. 4). It was verified via
direct DNA sequencing analysis that 166 bases in exon 7 were
completely deleted (Fig. 5). The
deletion results in an 8-base-frameshift from the 207th codon and a
premature termination at the 214th codon of the GHR mRNA. It
was considered to be a frame shifting change from methionine in
codon 207, with the new reading frame ending in a stop codon, M207
fs. X8.
Discussion
We report a 2-year-old patient with NF-1 who
presented postnatal growth failure with decreased growth velocity.
This patient exhibited very high GH concentrations after
stimulation with insulin and very low baseline levels of serum
IGF-1 and IGFBP3. The patient was ultimately diagnosed as having
GHIS through molecular analysis.
GHIS have been attributed to genetic defects along
the GH-IGF-1 axis. GH-IGF-1 axis plays a key role for body growth.
Therefore, GHIS shows the pathologic conditions associated with low
serum IGF-1 and IGFBP3 concentrations, as well as, normal or
elevated serum GH concentrations, leading to postnatal growth
failure including various clinical facial findings (8). An analysis that investigated growth
patterns in untreated patients with GHIS has revealed that most
patients showed reduced growth velocity and their growth charts
were similar to those with GH deficiency or IGF-1 gene deletion
(9).
A GH secretory problem was highly suspected as the
etiology of growth failure in our patient because the patient
demonstrated postnatal growth retardation and decreased height
growth velocity. In the present study, other hormonal secretory
functions were normal but serum GH concentrations remained very
high after stimulation with insulin. Also, the basal serum IGFBP3
and IGF-1 levels were too low to measure quantitatively. These
results reveal that the patient has GHIS and the growth failure
would be associated with it, not NF-1.
NF-1 related factors causing postnatal growth
failure were not found in the patient. The exact mechanism of
growth failure in NF-1 remains unclear, but it is known to be
associated with NF-1 itself or its complications (10). These complications include central
nervous system tumors, especially suprasellar lesions such as an
optic glioma, true precocious puberty, GH deficiency,
hypothyroidism, and abnormal skeletal development (11). In particular, GH deficiency and
true precocious puberty are the major risk factors of short stature
in NF-1 that are highly associated with central nervous system
pathology (2). No abnormal
findings were observed on MRI scanning of the
neuro-musculo-skeletal system in our patient. In addition, the
possibility of familial short stature as the etiology was excluded
because the heights of the patient’s family members were all within
normal limits.
It has been reported that in NF-1 patients during
puberty, growth spurt was slightly decreased and height according
to age gradually reduced, and eventually adult final heights in
NF-1 patients were less than those in normal individuals, whereas
growth velocity remained within normal range in prepubertal stage
(9,12). Because our patient showed
gradually reduced growth velocity since birth, the growth curve was
different from that of other NF-1 patients. Considering the reduced
growth velocity, low serum IGF-1 and IGFBP3 levels and abnormal
serum GH secretory pattern in the GH stimulation test, we strongly
suggested that the short stature of this patient is due to GHIS,
not NF-1.
Molecular studies were performed in order to
investigate whether the growth retardation in our patient is
associated with molecules involved in the intracellular signaling
cascade of the GH-IGF-1 axis. We found a novel mutation of
GHR mRNA, a complete deletion of exon 7, in the patient.
Because of the deletion and resulting frameshift, the mutation
results in a premature termination at the 214th codon (M207 fs. X8)
of the GHR gene. Exon 7 encodes a part of the extracellular
domain of the GHR protein (13,14). Therefore, the deletion mutation of
exon 7 is considered to make a truncated receptor protein, and then
the GH signal can not be transmitted into the cell. Reportedly,
almost all mutations causing Laron syndrome and GHIS encode the
extracellular domains of the GHR gene, while other mutations
encode the cytoplasmic domain (15,16).
In the reports regarding GHR mutation, mRNA
mutations of the GHR are uncommon, with splicing errors by
mutations of the flanking region of exon being the majority
(17). Rarely, mutations due to
addition of a pseudoexon in an intron were reported (18–20). The patient in the present study is
the first reported case having a GHIS with NF-1, who showed
extensive deletion of the GHR gene mRNA. To clarify the
mechanism of the disease-causing mutation, further studies are
required.
Acknowledgements
We thank the family for participation
in this study. This study was supported by the 2009 Eulji Research
Grant (EJRG-09-005-12E13).
References
|
1.
|
Neurofibromatosis Conference
statementNational Institutes of Health Consensus Development
ConferenceArch Neurol455755781988
|
|
2.
|
D CarmiM ShohatA MetzkerZ DickermanGrowth,
puberty, and endocrine functions in patients with sporadic or
familial neurofibromatosis type 1: a longitudinal
studyPediatrics10312571262199910.1542/peds.103.6.125710353939
|
|
3.
|
EM KofoedV HwaB LittleGrowth hormone
insensitivity associated with a STAT5b mutationN Engl J
Med34911391147200310.1056/NEJMoa02292613679528
|
|
4.
|
V HwaB LittleP AdiyamanSevere growth
hormone insensitivity resulting from total absence of signal
transducer and activator of transcription 5bJ Clin Endocrinol
Metab9042604266200510.1210/jc.2005-051515827093
|
|
5.
|
Z LaronA PertzelanS MannheimerGenetic
pituitary dwarfism with high serum concentration of growth hormone
- a new inborn error of metabolism?Isr J Med
Sci215215519665916640
|
|
6.
|
Z LaronLaron syndrome (primary growth
resistance or insensitivity): the personal experience 1958–2003J
Clin Endocrinol Metab8910311044200415001582
|
|
7.
|
F VidalE FarssacC AltisentL PuigD
GallardoRapid hemophilia A molecular diagnosis by a simple DNA
sequencing procedure: identification of 14 novel mutationsThromb
Haemost85580583200111341489
|
|
8.
|
PD GluckmanAJ GunnA WrayCongenital
idiopathic growth hormone deficiency is associated with prenatal
and early postnatal growth failureJ
Pediatr121920923199210.1016/S0022-3476(05)80342-71447657
|
|
9.
|
KA WoodC Cabacho-HubnerMO SavageAJ
ClarkIntrauterine growth retardation and postnatal growth failure
associated with deletion of insulin-like growth factor I geneN Eng
J Med33513631367199610.1056/NEJM1996103133518058857020
|
|
10.
|
MH CnossenEN StamLC CooimanEndocrinologic
disorders and optic pathway gliomas in children with
neurofibromatosis type
1Pediatrics100667670199710.1542/peds.100.4.6679310522
|
|
11.
|
J SzudeckP BirchJM FriedmanGrowth in North
American white children with neurofibromatosis 1 (NF1)J Med
Genet37933938200010.1136/jmg.37.12.933
|
|
12.
|
M ClementiS MilaniI MammiNeurofibromatosis
type 1 growth chartsAm J Med
Genet87317323199910.1002/(SICI)1096-8628(19991203)87:4%3C317::AID-AJMG7%3E3.0.CO;2-X10588837
|
|
13.
|
MA BergJ ArgenteS ChernausekDiverse growth
hormone receptor gene mutations in Laron syndromeAm J Hum
Genet52998100519938488849
|
|
14.
|
A EdensF TalamantesAlternative processing
of growth hormone receptor transcriptsEndocr
Rev1955958219989793757
|
|
15.
|
AL RosenbloomaJ Guevara-AguirrebLessons
from the genetics of Laron syndromeTrends Endocrinol
Metab9276283199810.1016/S1043-2760(98)00070-8
|
|
16.
|
PE ClaytonJS FreethMR NormanCongenital
growth hormone insensitivity syndromes and their relevance to
idiopathic short statureClin Endocrinol
(Oxf)50275283199910435051
|
|
17.
|
A DavidLA MetherellAJ ClarkC
Camacho-HübnerMO SavageDiagnostic and therapeutic advances in
growth hormone insensitivityEndocrinol Metab Clin North
Am34581595200510.1016/j.ecl.2005.04.00916085161
|
|
18.
|
LA MetherellSA AkkerPB MunroePseudoexon
activation as a novel mechanism for disease resulting in atypical
growth-hormone insensitivityAm J Hum
Genet69641646200110.1086/32326611468686
|
|
19.
|
M MaamraA MilwardHZ EsfahaniA 36 residues
insertion in the dimerization domain of the growth hormone receptor
results in defective trafficking rather than impaired signalingJ
Endocrinol188251261200610.1677/joe.1.0625216461551
|
|
20.
|
A DavidC Camacho-HübnerA BhangooAn
intronic growth hormone receptor mutation causing activation of a
pseudoexon is associated with a broad spectrum of growth hormone
insensitivity phenotypesJ Clin Endocrinol
Metab92655659200710.1210/jc.2006-1527
|