Introduction
The cardiac oxygen-sensing mechanism has attracted
extensive attention. A variety of genes relating to hypoxia
adaptation are induced at low-oxygen tensions, including
erythropoietin (EPO), vascular endothelial growth factor (VEGF),
glycolytic enzymes, glucose transporters, heme proteins, and
hypoxia inducible factor-1 (HIF-1) in various cell types (1–4).
The HIF-1 is the first identified family of transcription factors
that are rapidly elicited by hypoxia by inhibition of their
ubiquitin-dependent degradation (5). Previously, the important role of
HIF-1 in the cellular mechanism of oxygen-sensing was demonstrated
by confirming that the upregulation of 89% of genes induced by
hypoxia is HIF-1-dependent (6).
In their study, Baird et al (7) reported that heat shock proteins
(HSPs) are regulated by HIF-1 during hypoxia.
It has been well documented that cobalt chloride
(CoCl2) is a well-known hypoxia mimetic agent that
mimics the hypoxic response in various ways (3). CoCl2-mimicked hypoxia
enhances the level of HIF-1α protein (8–10).
Thus, CoCl2 has been used to explore the cardiac oxygen-sensing
mechanism in various experiment models. In isolated, perfused rat
hearts in hypoxia/reoxygenation, prior chronic oral
CoCl2 improves cardiac functions by increasing the
levels of VEGF, aldolase-A, and glucose transporter-1 (11). Furthermore, CoCl2
pretreatment before prolonged deep hypothermic circulatory arrest
(DHCA) rescues myocardial apoptosis by mechanisms involving the
phosphorylation of Akt, upregulation of the antiapoptotic protein
Bcl-2 expression and a decreased expression of the proapoptotic
protein Bax (12). These results
showed that preconditioning with CoCl2 protects against
cardiac injury induced by hypoxia or ischemia. The clinical
significance of such a strategy, however, is limited by the
unpredictability of hypoxia or ischemia time. The postconditioning
has been developed and has a similar effect as preconditioning in a
canine model of reversible coronary occlusion (13). Since the hypoxia and/or
ischemia-related diseases or injuries are clinically common,
developing a new therapeutic strategy for treatment of these
diseases or injuries is essential. Since cobalt has been
administered to human patients for the treatment of anemia
(14), and induces several
hypoxia-related protective factors, such as EPO (15,16), HSP90 (17) and heme oxgenase-1 (HO-1) (18,19), we speculates that the co-existence
of low concentrations of CoCl2 with serum and glucose
deprivation (SGD) is likely to attenuate ischemia-like injury in
cardiomyocytes.
To test this hypothesis, we undertook to examine the
protective effect of low concentrations of CoCl2 against
SGD-induced injury of H9c2 cardiomyocytes (H9c2 cells), as well as
the role of HSP90 in this cardioprotection and its mechanisms. The
findings of the present study have demonstrated that the
co-treatment of CoCl2 significantly protects H9c2 cells
against SGD-induced injury. Activation of the oxygen-sensing
mechanism by CoCl2 upregulates the expression of HSP90,
which is an important CoCl2-induced adaptive response to
the SGD condition. Such molecular adaptation to ischemia-like
events may contribute to the functional improvement of
cardiomyocytes, including antioxidation and the preservation of
MMP.
Materials and methods
Materials
CoCl2, glucose,
17-allylamino-17-demethoxygelda-namycin (17-AAG),
dichlorofluorescin diacetate (DCFH-DA) and Rhodamine 123 (Rh123)
were purchased from Sigma-Aldrich Corporation (St. Louis, MO, USA).
HSP90 antibody was purchased from Bioworld Technology (Minneapolis,
MN, USA). The cell counter kit-8 (CCK-8) was purchased from Dojindo
Laboratories (Kyushu, Japan). Glucose-free DMEM medium and fetal
bovine serum (FBS) were supplied by Gibco-BRL (Calsbad, CA,
USA).
Cell culture and treatments
H9c2 cells were obtained from Sun Yat-sen University
Experimental Animal Centre (Guangzhou, China). The H9c2 cell line,
a subclone of the original clonal cell line, was derived from
embryonic rat heart tissue. The cells were cultured in DMEM medium
supplemented with normal glucose (5.5 mM) and 15% FBS at 37°C under
an atmosphere of 5% CO2 and 95% air.
H9c2 cells were incubated in glucose- and FBS-free
DMEM medium to set up a model of SGD and mimic ischemia-induced
cardiac injury. CoCl2 was added into the above DMEM
medium to examine its cytoprotective effects. The selective HSP90
inhibitor, 17-AAG, was administered for 60 min prior to exposure of
the H9c2 cells to CoCl2 and SGD.
Cell viability assay
Cell viability was detected by the CCK-8 kit. H9c2
cells were cultured in 96-well plates at a density of 5,000
cells/well. When the cells were ∼70% fused, the indicated
treatments were performed. After the treatments, 10 μl CCK-8
solution was added into each well and the plates were incubated for
3 h. The absorbance at 450 nm was measured with a microplate reader
(Molecular Devices, Sunnyvale, CA, USA). A mean optical density
(OD) of 4 wells in each group was used to calculate percentage of
cell viability using the formula: percentage of cell viability = OD
treatment group/OD control group x100%. Experiments were performed
in triplicate.
Western blotting
At the end of the treatments, the H9c2 cells were
harvested and lysed with ice-cold cell lysis solution and the
homogenate was centrifuged at 10,000 x g for 12 min at 4°C. Total
protein in the supernatant was quantified using a BCA protein assay
kit. Total protein (20 μg) from each sample was separated by 12%
SDS-PAGE. The protein in the gel was transferred into
polyvinylidene difluoride (PVDF) membrane. The membrane was blocked
with 5% fat-free milk in TBS-T for 2 h at room temperature, and
then incubated with the primary antibodies specific to HSP90
(1:2,000), or β-actin (1:8,000) with gentle agitation at 4°C
overnight, followed by horseradish peroxidase-conjugated secondary
antibodies for 60 min at room temperature. After three washes with
TBS-T, the membranes were developed using an enhanced chemi
luminescence and exposed to X-ray film. To quantify the protein
expression, the X-ray film was scanned and analyzed using ImageJ
1.46i software (National Institutes of Health, USA).
Measurement of intracellular reactive
oxygen species (ROS) level
Intracellular ROS was determined by the oxidative
conversion of cell-permeable DCFH-DA to fluorescent DCF. H9c2 cells
were cultured on a slide in DMEM-F12 medium. After the indicated
treatments, the slides were washed twice with phosphate-buffered
saline (PBS). DCFH-DA solution in serum-free medium was added at a
concentration of 10 μM and co-incubated with H9c2 cells at 37°C for
60 min. The slides were washed three times, and DCF fluorescence
was measured over the entire field of vision using a fluorescent
microscope connected to an imaging system (BX50-FLA; Olympus). Mean
fluorescence intensity (MFI) from three random fields was analyzed
using ImageJ 1.46i software and the MFI of DCF represented the
amount of ROS.
Measurement of mitochondrial membrane
potential (MMP)
MMP was monitored with a fluorescent dye Rh123, a
cell-permeable cationic dye that preferentially enters into
mitochondria based on the highly negative MMP. Depolarization of
MMP results in the loss of Rh123 from mitochondria and a decrease
in intracellular green fluorescence. Rh123 (100 mg/l) was added
into cell cultures for 30 min at 37°C and fluorescence was measured
over the entire field of vision with a fluorescent microscope
connected to an imaging system (BX50-FLA; Olympus). MFI of Rh123
from three random fields was analyzed using ImageJ 1.46i software
and MFI was regarded as an index of the level of MMP.
Statistical analysis
Data were shown as the mean ± SE. The assessment of
differences between groups was analyzed by Tukey’s test of one-way
ANOVA with OriginPro 8.0 (OriginLab Corporation, Northampton, MA,
USA). P<0.05 was considered to indicate statistical
significance.
Results
SGD time-dependently enhances
cytotoxicity in H9c2 cells
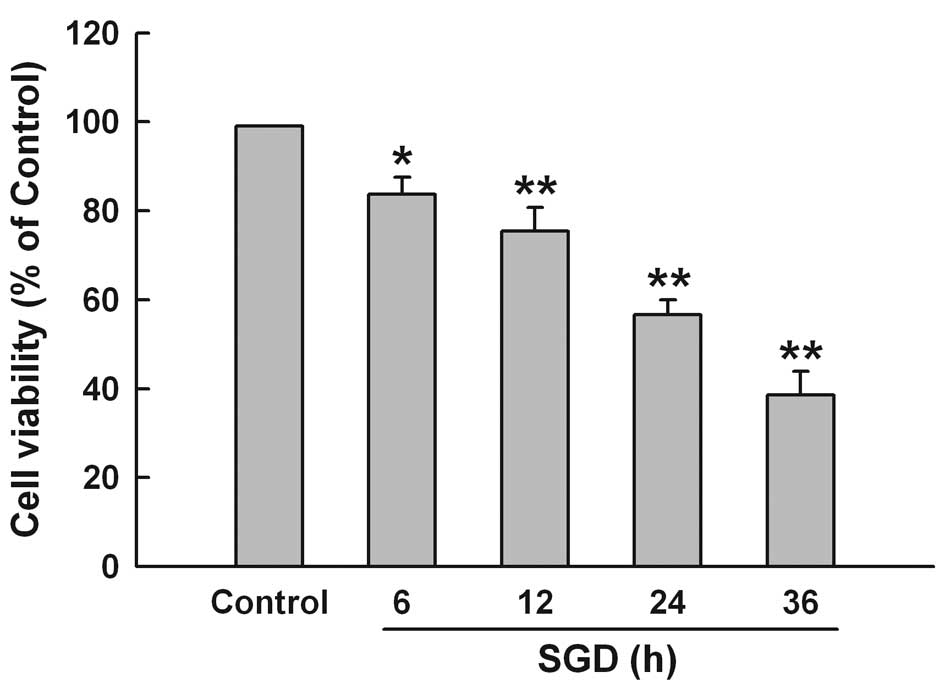
To test the effect of SGD on cytotoxicity in H9c2
cells, the CCK-8 assay was performed. Fig. 1 shows that SGD obviously induced
cytotoxicity, leading to a time-dependent decrease in cell
viability. Cell survival was decreased to 56.7±3.3% by 24 h of
incubation in serum- and glucose-free medium. Therefore, 24 h of
SGD was used as an effective injury time in the subsequent
experiments.
SGD downregulates the expression of HSP90
in H9c2 cells
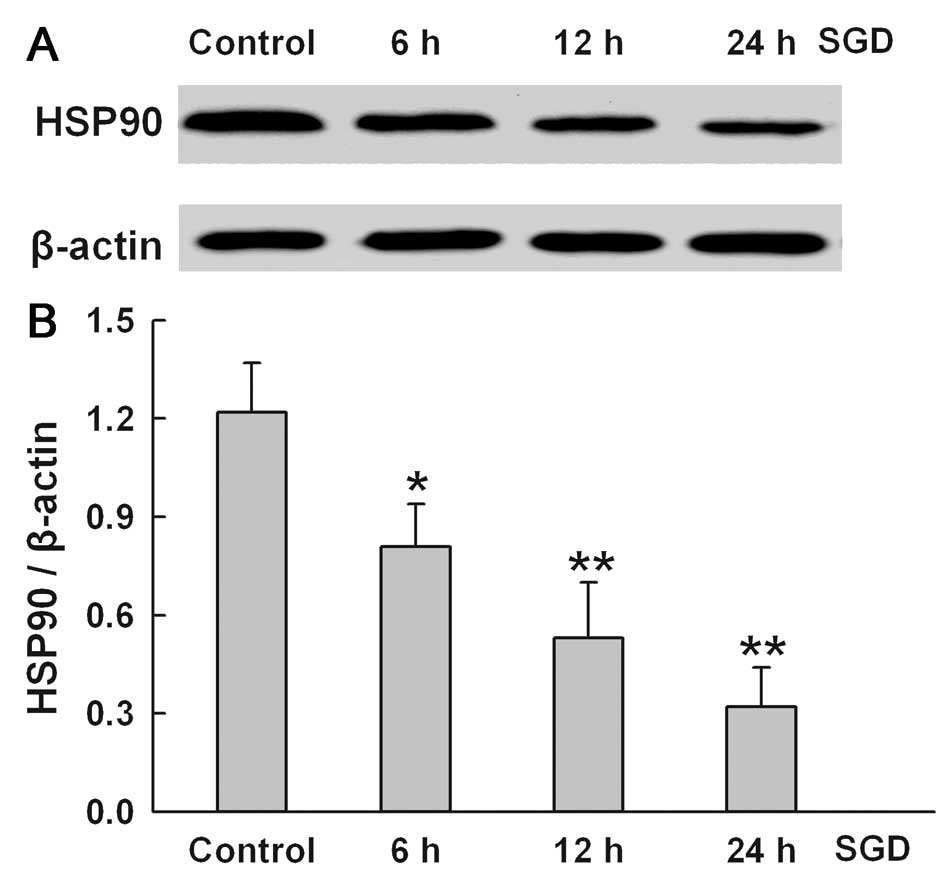
To determine the effect of SGD on HSP90 expression,
the changes in HSP90 expression were tested in serum- and
glucose-deprived H9c2 cells for the indicated times (6, 12 and 24
h), respectively. SGD attenuated HSP90 expression in a
time-dependent manner, suggesting that the downregulation of HSP90
expression may be one of the mechanisms involved in SGD-induced
injury in the H9c2 cells (Fig.
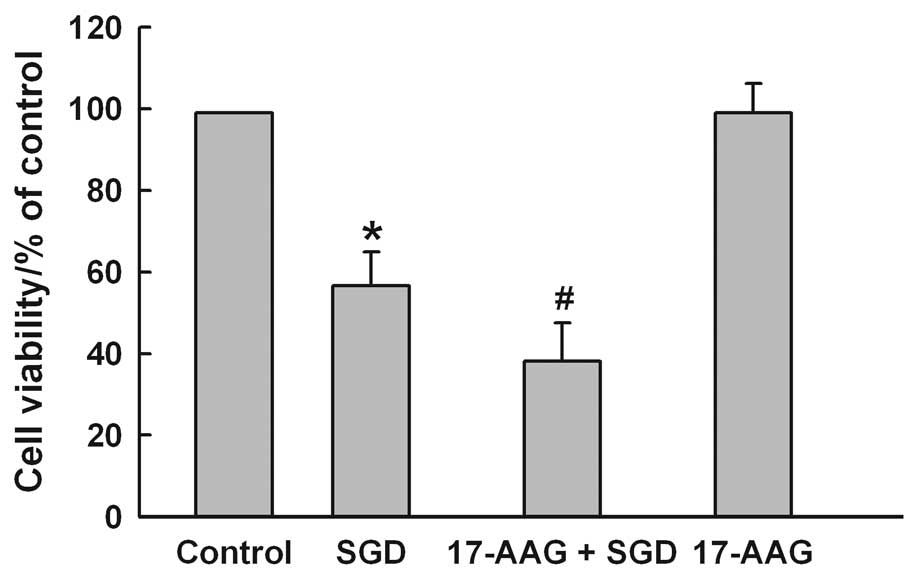
2). The results of subsequent experiments showed that
pretreatment of the cells with 2 μM 17-AAG, a selective inhibitor
of HSP90, for 60 min before SGD for 24 h markedly aggravated
SGD-induced cytotoxicity, evidenced by a further decrease in cell
viability (Fig. 3). However,
17-AAG alone did not alter cell viability in H9c2 cells. These
results showed that HSP90 may be one of the molecular defensive
mechanisms against SGD-induced injury in H9c2 cells.
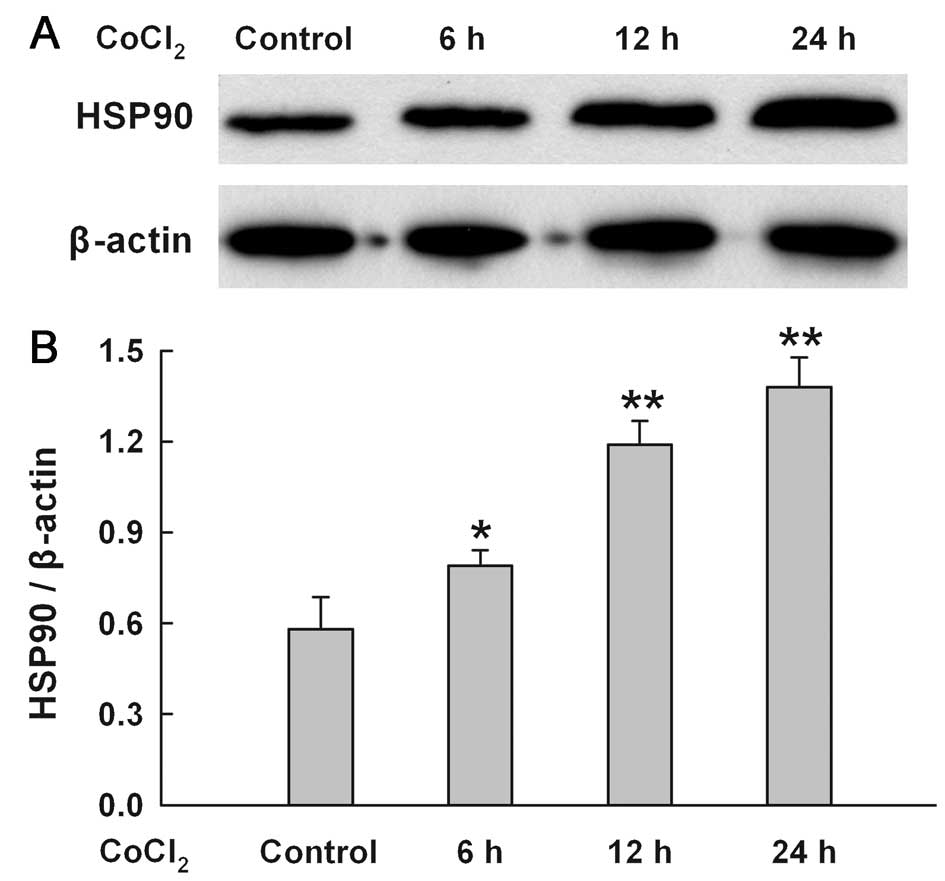
CoCl2 time-dependently
upregulates the expression of HSP90 in H9c2 cells
As shown in Fig.
4, when H9c2 cells were treated with 100 μM CoCl2
for 6–24 h, a time-dependent increase in HSP90 expression was
found.
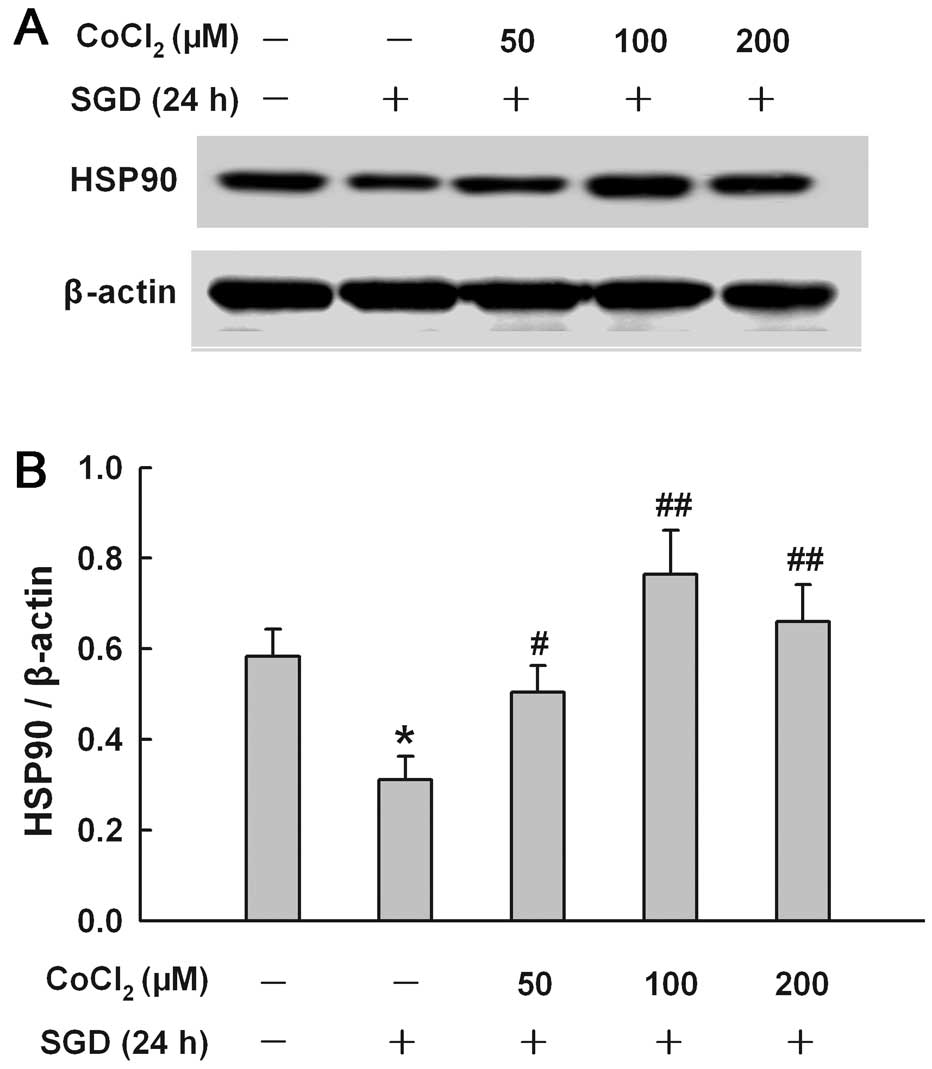
CoCl2 reduces the inhibitory
effect of SGD on the expression of HSP90 in H9c2 cells
To explore the effect of chemical hypoxia on the
SGD-induced inhibition of HSP90 expression, H9c2 cells were treated
with CoCl2 at concentrations ranging from 50 to 200 μM
for 24 h in the presence of SGD, respectively. Fig. 5 shows that co-treatment with
CoCl2 at 50, 100 and 200 μM inhibited the downregulation
of the SGD-induced HSP90 expression. A maximal inhibitory effect of
CoCl2 was observed at 100 μM.
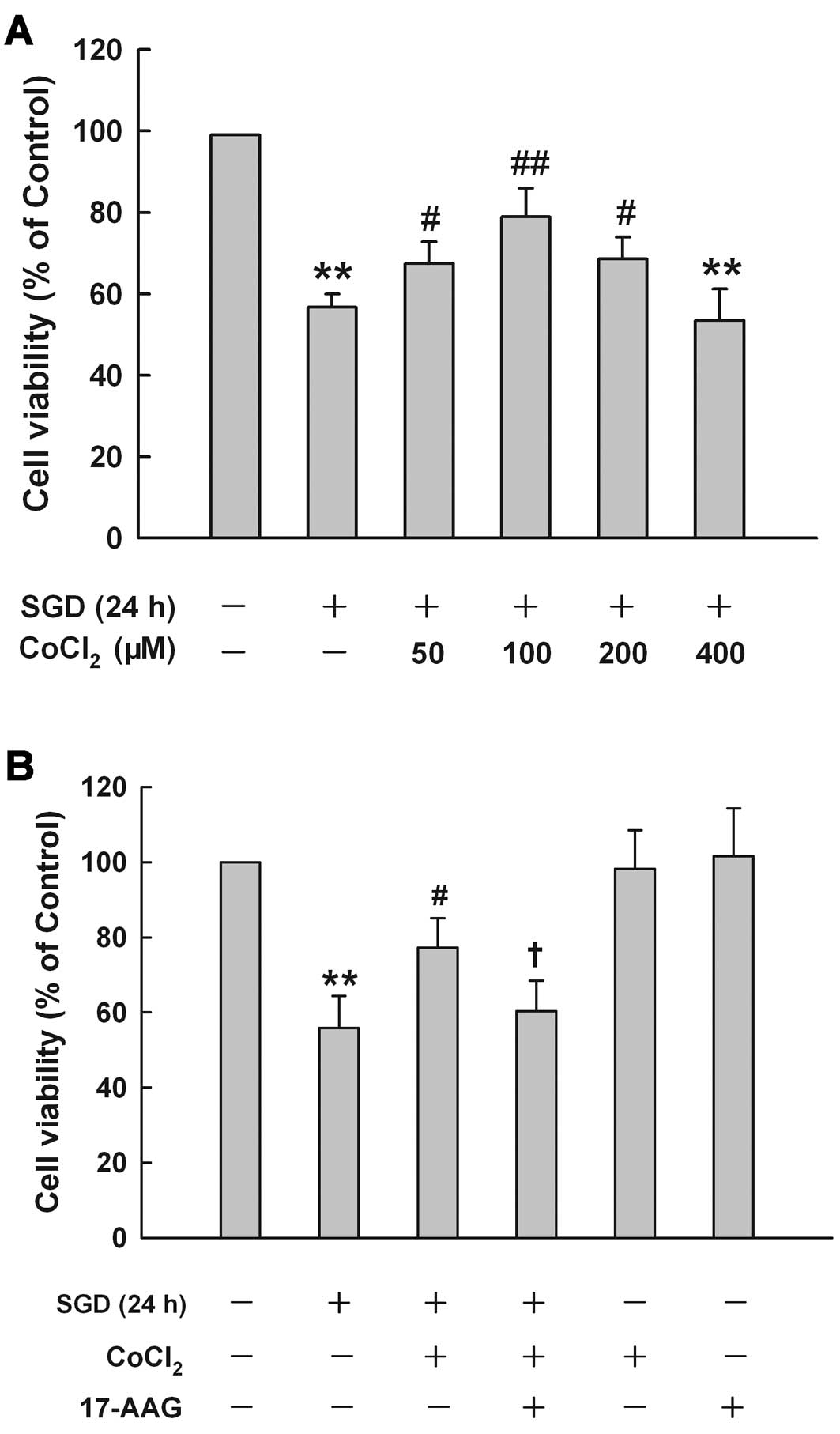
HSP90 contributes to the cytoprotection
at low concentrations of CoCl2 against SGD-induced
cytotoxicity in H9c2 cells
CoCl2 at different concentrations had
different effects on SGD-induced cytotoxicity in H9c2 cells
(Fig. 6A). Treatment of cells
with low concentrations (50, 100 and 200 μM) obviously protected
against SGD-induced cytotoxicity, leading to an increase in cell
viability. These concentrations demonsrated a maximum
cytoprotective effect of CoCl2 at 100 μM, which alone
did not affect cell viability (data not shown). Therefore, 100 μM
of CoCl2 was used as an effective protection
concentration in the subsequent experiments. These results suggest
that sublethal chemical hypoxia treatment is protective against
ischemia-like injury.
To investigate the role of HSP90 in the
cytoprotective effect of CoCl2 at low concentrations
against SGD-induced injury, H9c2 cells were pretreated with 2 μM
17-AAG, a selective inhibitor of HSP90, for 60 min prior to the
co-treatment of 100 μM CoCl2 with SGD for 24 h. Fig. 6B shows that cell treatment with
100 μM CoCl2 considerably reduced SGD-induced
cytotoxicity, evidenced by an increase in cell viability. However,
this cytoprotection of CoCl2 treatment was reduced by
17-AAG pretreatment, suggesting that HSP90 is involved in the
protective effect of sublethal chemical hypoxia against cardiac
toxicity induced by SGD in H9c2 cells. However, 2 μM 17-AAG alone
did not alter cell viability.
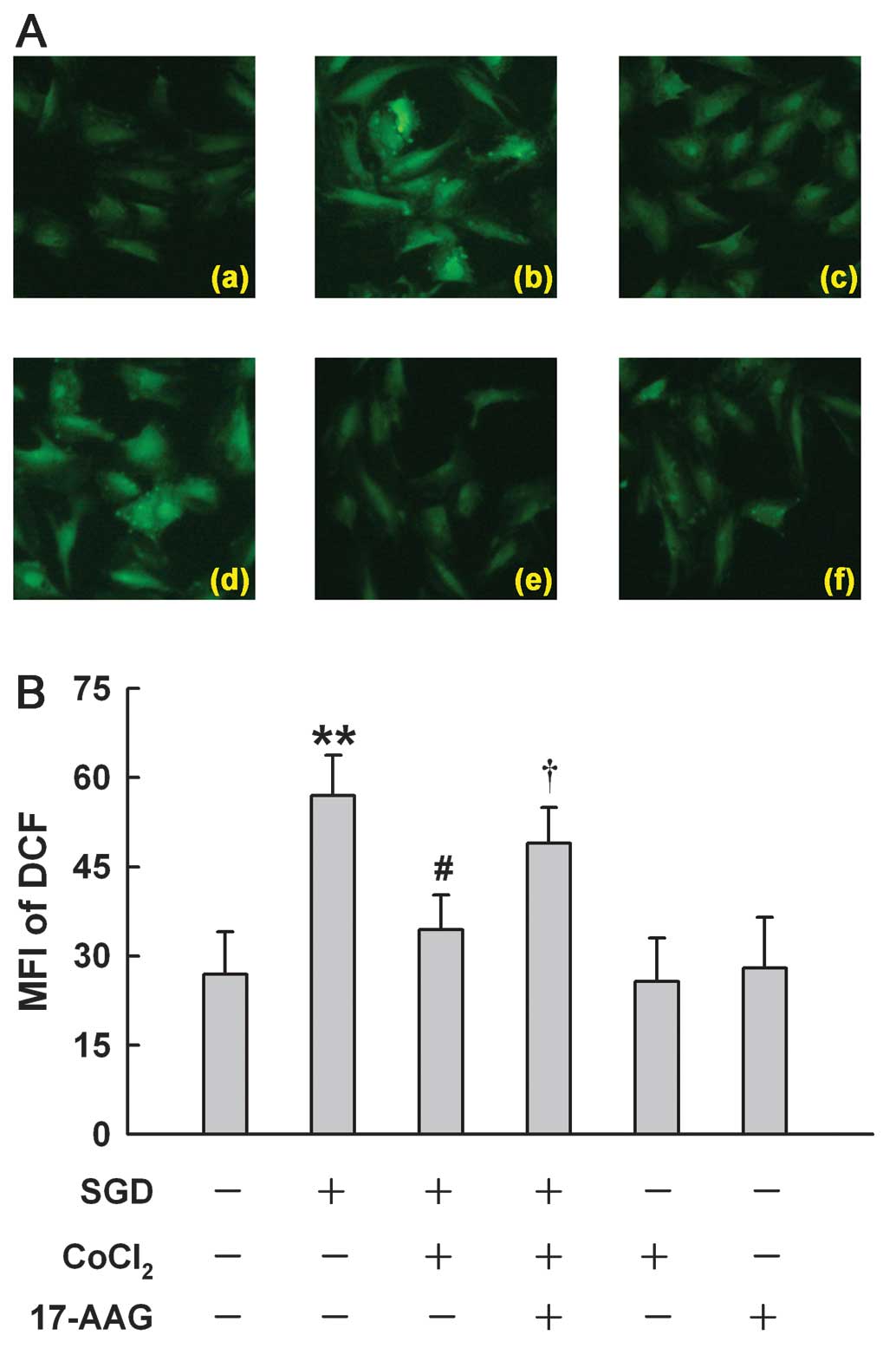
HSP90 participates in the antioxidant
effect induced by a low concentration of CoCl2 in H9c2
cells
A more recent study showed that a high concentration
(600 μM) of CoCl2 induces ROS generation in H9c2 cells
(17). Of note, we found that
CoCl2 at 100 μM reduced SGD-induced ROS generation,
manifesting a decrease in DCF-derive fluorescence (an index of ROS
level) (Fig. 7Ac). However, the
antioxidant effect of 100 M CoCl2 was markedly
suppressed by the inhibition of HSP90 with 17-AAG at 2 μM (Fig. 7Ad). Alone, 100 μM CoCl2
or 2 μM 17-AAG did not alter the basal level of ROS. These results
indicated that the antioxidant effect of low concentrations of
CoCl2 may be associated with an upregulated HSP90
expression.
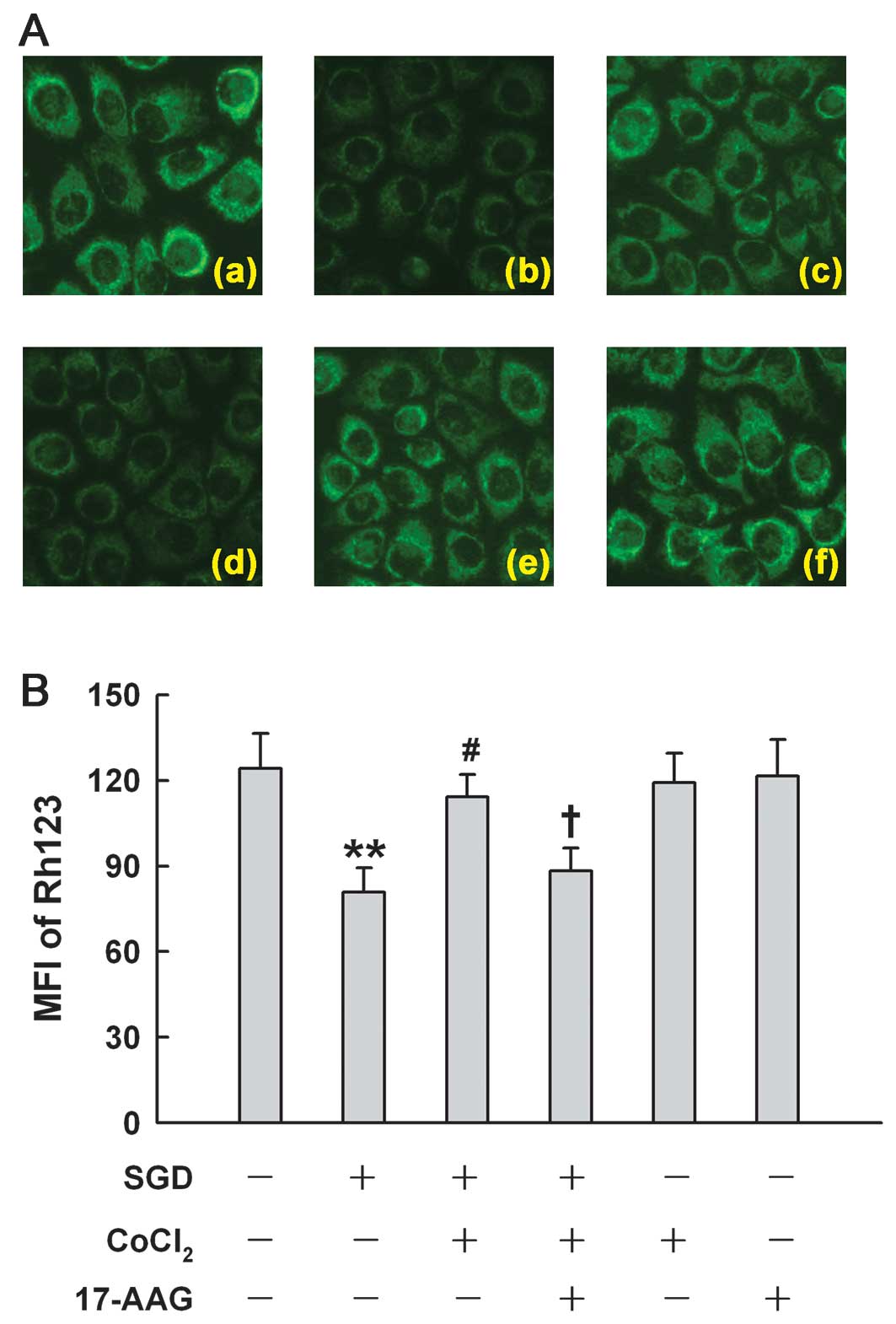
HSP90 is associated with the
mitochondrial protection induced by low concentrations of
CoCl2 in H9c2 cells
Fig. 8 shows that
after H9c2 cells were subjected to SGD for 24 h, mitochondria were
markedly damaged, leading to a decrease in the uptake of Rh123,
demonstrating dissipation of MMP (Fig. 8Ab). The loss of MMP was reduced by
treatment with 100 μM CoCl2 for 24 h (Fig. 8Ac). The inhibition of HSP90 by
17-AAG (2 μM) pretreatment significantly depressed the
mitochondrial protection induced by CoCl2, resulting in
the severe loss of MMP, suggesting that low concentrations of
CoCl2 provide mitochondrial protection by enhancing
HSP90 expression.
Discussion
The important findings of this study are that
co-treatment of low concentrations of CoCl2 with SGD
provides cardioprotection against ischemia-like injury in H9c2
cells and that HSP90 contributes to the cardioprotective effect of
sublethal chemical hypoxia from SGD-induced injury through its
antioxidation and mitochondrial protection. The present study
provides novel evidence to support the hypothesis that sublethal
endogenous hypoxia may be one of the defensive mechanisms by which
cardiomyocytes become more resistant to ischemia-induced injury and
that cobalt, a traditional medicine, may have therapeutic efficacy
for the treatment of ischemia-related cardiac diseases.
Previous studies have shown that pretreatment with
CoCl2, a well-known hypoxia mimetic agent, offers
cardioprotective effects, including attenuation of myocardial
apoptosis (12) and improvement
of cardiac contractile functions (11). Consistent with these studies
(11,12), in the present study, we found that
co-treatment of 100 μM CoCl2, which does not damage H9c2
cells, is able to protect against SGD-induced injury, leading to an
increase in cell viability, a decrease in ROS generation and a
preservation of MMP. Due to the unpredictability of hypoxia or
ischemia timing, the clinical significance of hypoxia
preconditioning is limited. Since co-treatment with
CoCl2 has a similar cardioprotection as CoCl2
preconditioning, the administration of cobalt used in this study
has potential clinical significance for the treatment of
ischemia-related heart diseases. Furthermore, our results provide
new data to demonstrate the inhibitory effect of cobalt on
ischemia-like cardiac injury.
Based on our findings and those of previous studies
(11,12,15–19), several possible mechanisms are
responsible for the cardioprotective effect of low concentrations
of CoCl2: i) its antioxidant effect, by which
CoCl2 is able to reduce SGD-induced ROS generation in
H9c2 cells. Consistent with our findings, a previous study has
shown that manganese-containing superoxide dismutase (SOD) and
mitochondrial-targeted catalase significantly protects against
cytotoxicity and the glucose deprivation-induced oxidative stress
(20); ii) its mitochondrial
protective effect, by which CoCl2 inhibits the
SGD-induced dissipation of MMP. Similarly, cobalt has been shown to
have direct action in preserving ATP in adult rat myocardium
(21); iii) its enhancement
effect on HIF-1 expression, by which CoCl2 is capable of
protecting isolated mouse hearts against ischemia/reperfusion
(I/R)-induced injury (22); iv)
its upregulation effect on EPO, which is one of the target products
of HIF-1. Cai et al (23)
reported that hearts exposed to intermittent hypoxia or EPO are
protected from I/R-induced injury. Previous studies have
demonstrated the effectiveness of EPO in limiting apoptosis in rat
cardiac myocytes (24,25); v) its enhancing the levels of
VEGF, by which low concentrations of CoCl2 improves
cardiac contractile functions (11). VEGF is a potent angiogenic factor
and its upregulation may facilitate tissue perfusion and
oxygenation in hypoxia, an important adaptation to hypoxia; vi) its
inductive effect of glucose transporters (GLUT). In the in
vivo heart and cultured cardiac cells, both hypoxia and
Co2+ rapidly induce GLUT-1 expression in the mRNA level
(26,27). The increased GLUT-1 is involved in
improved cardiac contractile function in hypoxia-reoxygenation in
rats treated with low concentrations of CoCl2 (11).
Besides the above six possible mechanisms underlying
the cardioprotection of low concentrations of CoCl2,
another important mechanism involves the activation of the
oxygen-sensing mechanism by CoCl2, which may upregulate
the expression of HSPs. HSPs are a family of protective proteins,
which provide an endogenous cell defense mechanism against hostile
environmental stress, including hypoxia and ischemia. Accumulating
evidence has shown that the upregulation of HSP expression is
capable of protecting cardiomyocytes from I/R-induced damage
(28) or chemical
hypoxia-triggered injury (17).
Notably, CoCl2 is also able to upregulate the expression
of HSP90 in H9c2 cells (17).
Based on these previous studies (17,29), we hypothesizes that HSP90
possibley contributes to the cardioprotective effect of
co-treatment with low concentrations of CoCl2 from
SGD-induced cardiomyocyte injury. In the present study, we
initially observed that SGD downregulated the expression of HSP90
and that 17-AAG, a selective inhibitor of HSP90, significantly
aggravated SGD-induced cytotoxicity in H9c2 cells, suggesting that
HSP90 is an endogenous protective protein against SGD-induced
injury. Secondly, we found that co-treatment with 100 μM
CoCl2, not only enhanced the expression of HSP90, but
also obviously blocked the inhibitory effect of SGD on HSP90
expression. Thirdly, examination into the roles of HSP90 in the
cardioprotection of low concentrations of CoCl2 against
SGD-induced injuries, including cytotoxicity, overproduction of ROS
and dysfunction of mitochondria was conducted. The findings of the
present study have shown that the inhibition of HSP90 by 17-AAG
markedly blocked the reduction of cytotoxicity induced by
CoCl2 at 100 μM, indicating the involvement of HSP90 in
the cardioprotective effect of CoCl2 against
SGD-triggered injury. Our results are comparable with those of a
recent study whereby the inhibition of HSP90 by geldanamycin, an
inhibitor of HSP90, or siRNA completely eliminates the protective
effect of hypoxic pretreatment against prolonged
hypoxia/reoxygenation-elicited damage in H9c2 cells (30). Yang et al (17) also reported that increased HSP90
expression participates in the cardioprotection induced by hydrogen
sulfide (H2S).
ROS is regarded as a toxic byproduct of aerobic
metabolism following reoxygenation after periods of ischemia or
hypoxia (31). Ahmad et al
(20) indicated that
mitochondrial ROS mediate glucose deprivation-induced cytotoxicity.
In this study, it was shown that co-treatment with low
concentrations of CoCl2 attenuated cytotoxicity, along
with a decrease in ROS production, suggesting that co-treatment
with CoCl2 is likely to induce antioxidants to reduce
ROS generation. Findings of a more recent study have demonstrated
the involvement of HSP90 in the antioxidant effect of
H2S (17). In
agreement with this evidence, our data showed that 17-AAG
significantly depressed CoCl2-induced inhibitory effect
on the overproduction of ROS elicited by SGD in H9c2 cells,
demonstrating that HSP90 is involved in the antioxidant effect of
CoCl2.
Mitochondria play a critical role in determining the
cell fate (32) by regulating the
mitochondrial permeability transition pore (PTP) and the release of
a number of apoptotic factors. Cell death inhibition is accompanied
by MMP preservation (17,33–35). Since antioxidants are able to
inhibit the loss of MMP, the role of HSP90 in the
CoCl2-induced cardioprotective effect may be associated
with the preservation of mitochondrial function. In this study,
co-treatment with CoCl2 considerably inhibited
SGD-induced loss of MMP, and this mitochondrial protective function
was blocked by the inhibition of HSP90 by 17-AAG. Our findings are
supported by those of Yang et al (17) and are consistent with a recent
finding that HSP90 is required for mitochondrial protein import for
hydrophobic membrane proteins, especially under stress conditions
(36).
Taken together, in the present study, we have
demonstrated that chemical hypoxia-upregulated HSP90 expression
contributes to the CoCl2-induced cardioprotective effect
from SGD-triggered injury by depressing cytotoxicity and oxidative
stress as well as preserving MMP. However, more experiments are
required to elucidate the activation of the cardiac oxygen-sensing
mechanism by CoCl2, in particular, the interaction
between HIF-1 and HSP90.
Acknowledgements
The present study was supported by the
Science and Technology Planning Project of Guangdong in China (nos.
2011B031800002 and 2011B080701051) and the Guangdong Natural
Science Foundation (no. S2011010002620).
References
|
1.
|
J FandreyHypoxia-inducible gene
expressionRespir Physiol101110199510.1016/0034-5687(95)00013-4
|
|
2.
|
JM GleadlePJ RatcliffeHypoxia and the
regulation of gene expressionMol Med
Today4122129199810.1016/S1357-4310(97)01198-29575495
|
|
3.
|
MA GoldbergSP DunningHF BunnRegulation of
the erythropoietin gene: evidence that the oxygen sensor is a heme
proteinScience24214121415198810.1126/science.28492062849206
|
|
4.
|
GL SemenzaGL WangA nuclear factor induced
by hypoxia via de novo protein synthesis binds to the human
erythropoietin gene enhancer at a site required for transcriptional
activationMol Cell Biol125447545419921448077
|
|
5.
|
GL SemenzaO2-regulated gene
expression: transcriptional control of cardiorespiratory physiology
by HIF-1J Appl Physiol96117311772004
|
|
6.
|
AE GreijerP van der GroepD
KemmingUp-regulation of gene expression by hypoxia is mediated
predominantly by hypoxia-inducible factor 1 (HIF-1)J
Pathol206291304200510.1002/path.177815906272
|
|
7.
|
NA BairdDW TurnbullEA JohnsonInduction of
the heat shock pathway during hypoxia requires regulation of heat
shock factor by hypoxia-inducible factor-1J Biol
Chem2813867538681200610.1074/jbc.M60801320017040902
|
|
8.
|
Y HuangKM DuZH XueCobalt chloride and low
oxygen tension trigger differentiation of acute myeloid leukemic
cells: possible mediation of hypoxia-inducible
factor-1alphaLeukemia1720652073200310.1038/sj.leu.2403141
|
|
9.
|
EJ KimYG YooWK YangTranscriptional
activation of HIF-1 by RORalpha and its role in hypoxia
signalingArterioscler Thromb Vasc
Biol2817961802200810.1161/ATVBAHA.108.17154618658046
|
|
10.
|
A LanX LiaoL MoHydrogen sulfide protects
against chemical hypoxia-induced injury by inhibiting ROS-activated
ERK1/2 and p38MAPK signaling pathways in PC12 cellsPLoS
One6e25921201110.1371/journal.pone.002592121998720
|
|
11.
|
H EndohT KanekoH NakamuraImproved cardiac
contractile functions in hypoxia-reoxygenation in rats treated with
low concentration Co(2+)Am J Physiol Heart Circ
Physiol279H2713H2719200011087225
|
|
12.
|
F KerendiPM KirshbomME HalkosThoracic
Surgery Directors Association Award. Cobalt chloride pretreatment
attenuates myocardial apoptosis after hypothermic circulatory
arrestAnn Thorac
Surg8120552062200610.1016/j.athoracsur.2006.01.059
|
|
13.
|
ZQ ZhaoJS CorveraME HalkosInhibition of
myocardial injury by ischemic postconditioning during reperfusion:
comparison with ischemic preconditioningAm J Physiol Heart Circ
Physiol285H579H588200310.1152/ajpheart.01064.200212860564
|
|
14.
|
JM DuckhamHA LeeThe treatment of
refractory anaemia of chronic renal failure with cobalt chlorideQ J
Med452772941976940922
|
|
15.
|
JW FisherJW LangstonEffects of
testosterone, cobalt and hypoxia on erythropoietin production in
the isolated perfused dog kidneyAnn NY Acad
Sci1497587196810.1111/j.1749-6632.1968.tb15139.x5240755
|
|
16.
|
WE JandaW FriedCW GurneyCombined effect of
cobalt and testosterone on erythropoiesisProc Soc Exp Biol
Med120443446196510.3181/00379727-120-305585856423
|
|
17.
|
Z YangC YangL XiaoNovel insights into the
role of HSP90 in cytoprotection of H2S against chemical
hypoxia-induced injury in H9c2 cardiac myocytesInt J Mol
Med28397403201121519787
|
|
18.
|
R Eyssen-HernandezA LadouxC
FrelinDifferential regulation of cardiac heme oxygenase-1 and
vascular endothelial growth factor mRNA expressions by hemin, heavy
metals, heat shock and anoxiaFEBS
Lett382229233199610.1016/0014-5793(96)00127-58605975
|
|
19.
|
D KatayoseS IsoyamaH FujitaSeparate
regulation of heme oxygenase and heat shock protein 70 mRNA
expression in the rat heart by hemodynamic stressBiochem Biophys
Res Commun191587594199310.1006/bbrc.1993.12588461015
|
|
20.
|
IM AhmadN Aykin-BurnsJE SimMitochondrial
O2*-and H2O2 mediate
glucose deprivation-induced stress in human cancer cellsJ Biol
Chem28042544263200515561720
|
|
21.
|
CH ConradWW BrooksJS IngwallInhibition of
hypoxic myocardial contracture by Cobalt in the ratJ Mol Cell
Cardiol16345354198410.1016/S0022-2828(84)80605-76726823
|
|
22.
|
L XiM TaherC YinCobalt chloride induces
delayed cardiac preconditioning in mice through selective
activation of HIF-1alpha and AP-1 and iNOS signalingAm J Physiol
Heart Circ
Physiol287H2369H2375200410.1152/ajpheart.00422.200415284066
|
|
23.
|
Z CaiDJ ManaloG WeiHearts from rodents
exposed to intermittent hypoxia or erythropoietin are protected
against ischemia-reperfusion
injuryCirculation1087985200310.1161/01.CIR.0000078635.89229.8A12796124
|
|
24.
|
CJ ParsaA MatsumotoJ KimA novel protective
effect of erythropoietin in the infarcted heartJ Clin
Invest1129991007200310.1172/JCI1820014523037
|
|
25.
|
L CalvilloR LatiniJ KajsturaRecombinant
human erythropoietin protects the myocardium from
ischemia-reperfusion injury and promotes beneficial remodelingProc
Natl Acad Sci USA10048024806200310.1073/pnas.063044410012663857
|
|
26.
|
WI SivitzDD LundB YorekPretranslational
regulation of two cardiac glucose transporters in rats exposed to
hypobaric hypoxiaAm J Physiol263E562E56919921415537
|
|
27.
|
J YbarraA BehroozA
GabrielGlycemia-lowering effect of cobalt chloride in the diabetic
rat: increased GLUT1 mRNA expressionMol Cell
Endocrinol133151160199710.1016/S0303-7207(97)00162-79406861
|
|
28.
|
C YinL XiX WangSilencing heat shock factor
1 by small interfering RNA abrogates heat shock-induced
cardioprotection against ischemia-reperfusion injury in miceJ Mol
Cell Cardiol39681689200510.1016/j.yjmcc.2005.06.005
|
|
29.
|
NA WhitlockN AgarwalJX MaHSP27
upregulation by HIF-1 signaling offers protection against retinal
ischemia in ratsInvest Ophthalmol Vis
Sci4610921098200510.1167/iovs.04-004315728570
|
|
30.
|
JD JiaoV GargB YangNovel functional role
of heat shock protein 90 in ATP-sensitive K+
channel-mediated hypoxic preconditioningCardiovasc
Res77126133200810.1093/cvr/cvm02818006464
|
|
31.
|
JM McCordOxygen-derived free radicals in
postischemic tissue injuryN Engl J
Med312159163198510.1056/NEJM1985011731203052981404
|
|
32.
|
DR GreenJC ReedMitochondria and
apoptosisScience28113091312199810.1126/science.281.5381.13099721092
|
|
33.
|
J ChenXQ TangJL ZhiCurcumin protects PC12
cells against 1-methyl-4-phenylpyridinium ion-induced apoptosis by
Bcl-2-mitochondria-ROS-iNOS
pathwayApoptosis11943953200610.1007/s10495-006-6715-516547587
|
|
34.
|
K ZhaoGM ZhaoD WuCell-permeable peptide
antioxidants targeted to inner mitochondrial membrane inhibit
mitochondrial swelling, oxidative cell death, and reperfusion
injuryJ Biol Chem2793468234690200410.1074/jbc.M402999200
|
|
35.
|
SL ChenCT YangZL YangHydrogen sulphide
protects H9c2 cells against chemical hypoxia-induced injuryClin Exp
Pharmacol
Physiol37316321201010.1111/j.1440-1681.2009.05289.x19769612
|
|
36.
|
JC YoungNJ HoogenraadFU HartlMolecular
chaperones Hsp90 and Hsp70 deliver preproteins to the mitochondrial
import receptor
Tom70Cell1124150200310.1016/S0092-8674(02)01250-312526792
|