Introduction
Traumatic joint damage is a major risk factor for
development and/or progression of osteoarthritis (OA) (1). Knee injuries often result from
traffic accidents and count for 40% of sports injuries (2–4).
The high prevalence of post-traumatic arthritis creates the urgent
need for pharmacological interventions, starting in the early phase
after injury in which the destructive processes are induced
(1,5). Therefore, a detailed knowledge of
the underlying pathogenetic mechanisms is crucial but only partly
available. The excessive mechanical loading of the articular
cartilage results in chondrocyte death that can be attributed to
mechanical necrosis (6) and
apoptotic processes (7). In
addition, mechanical disruption of the extracellular matrix leads
to an early loss of glycosaminoglycans (8). Cartilage defects in knee injuries
are commonly associated with other pathological conditions of the
joint such as anterior cruciate ligament (ACL) ruptures and
meniscal injuries (9), leading to
an increase of proinflammatory cytokines in the synovial fluid
(10). Beside IL-1β and IL-6,
which are elevated during the first 3 to 6 days, the level of TNF-α
is significantly increased over a period of 3 weeks after ACL
ligament injury of the knee with a peak concentration in the first
24 h (11).
Proinflammatory cytokines are known to drive the
overall inflammatory process by inducing other cytokines,
chemokines and proinflammatory mediators like prostaglandins and NO
(12,13). These cytokines enhance cartilage
degradation via an activation of catabolic and a decrease of
anabolic processes. In particular, degenerative enzymes like matrix
metalloproteinases (MMP) 1, 3 and 13 as well as aggrecanases (e.g.
ADAMTS 4/5) are induced by TNF-α and/or IL-1β (14–16). In vitro studies with human
and bovine cartilage revealed that TNF-α and IL-1α potentiate the
effect of cartilage impact, leading to a synergistic loss of
proteoglycans and to a reduction of their biosynthesis (17,18). In contrast, IL-1β co-stimulation
had no additive effects on cell death in traumatized cartilage
during the first 24 h but prostaglandin (PGE2 and PGD2) and NO
synthesis pathways were both affected by trauma and/or IL-1β
(19). In an apoptosis model of
chondrocytes sensitized by actinomycin D, TNF-α but not IL-1β was
described to enhance apoptosis indicating differential regulations
by these cytokines (20,21). So far, no studies exist on
interactions of TNF-α and a single impact cartilage trauma with
respect to cell death, prostaglandin or NO metabolism.
Besides well-known effects on cartilage degeneration
and inflammation in arthritic diseases like induction of MMPs,
aggrecanases, inducible nitric oxide synthase and cyclooxygenase
(22,23), TNF-α is also involved in the
regulation of bone remodeling by stimulation of receptor activator
of nuclear factor (NF-κB) ligand (RANKL) expression in osteoblasts.
Binding of RANKL to its receptor RANK on osteoclasts increases bone
resorption by promoting their differentiation and activation
(24). RANKL and its decoy
receptor osteoprotegerin (OPG) are also expressed in human
chondrocytes and may play a role in osteoarthritis, where OPG
production is increased (25).
They may alter the remodeling process of subchondral bone which
becomes sclerotic during development of post-traumatic OA.
Overall, TNF-α could have significant influence in
the early phase after cartilage trauma. A deeper knowledge of its
contribution to induction or progression of post-traumatic OA could
be directive for new therapeutic approaches. Therefore, the primary
aims of the present study were to elucidate, whether TNF-α
potentiates chondrocyte death, alters the release of
proinflammatory mediators (PGE2, PGD2, NO and IL-6) and modulates
OPG/RANKL expression in human cartilage in the initial phase after
a defined single impact trauma. We further asked if these
treatments have early effects on gene expression of catabolic
enzymes and cartilage matrix components.
Materials and methods
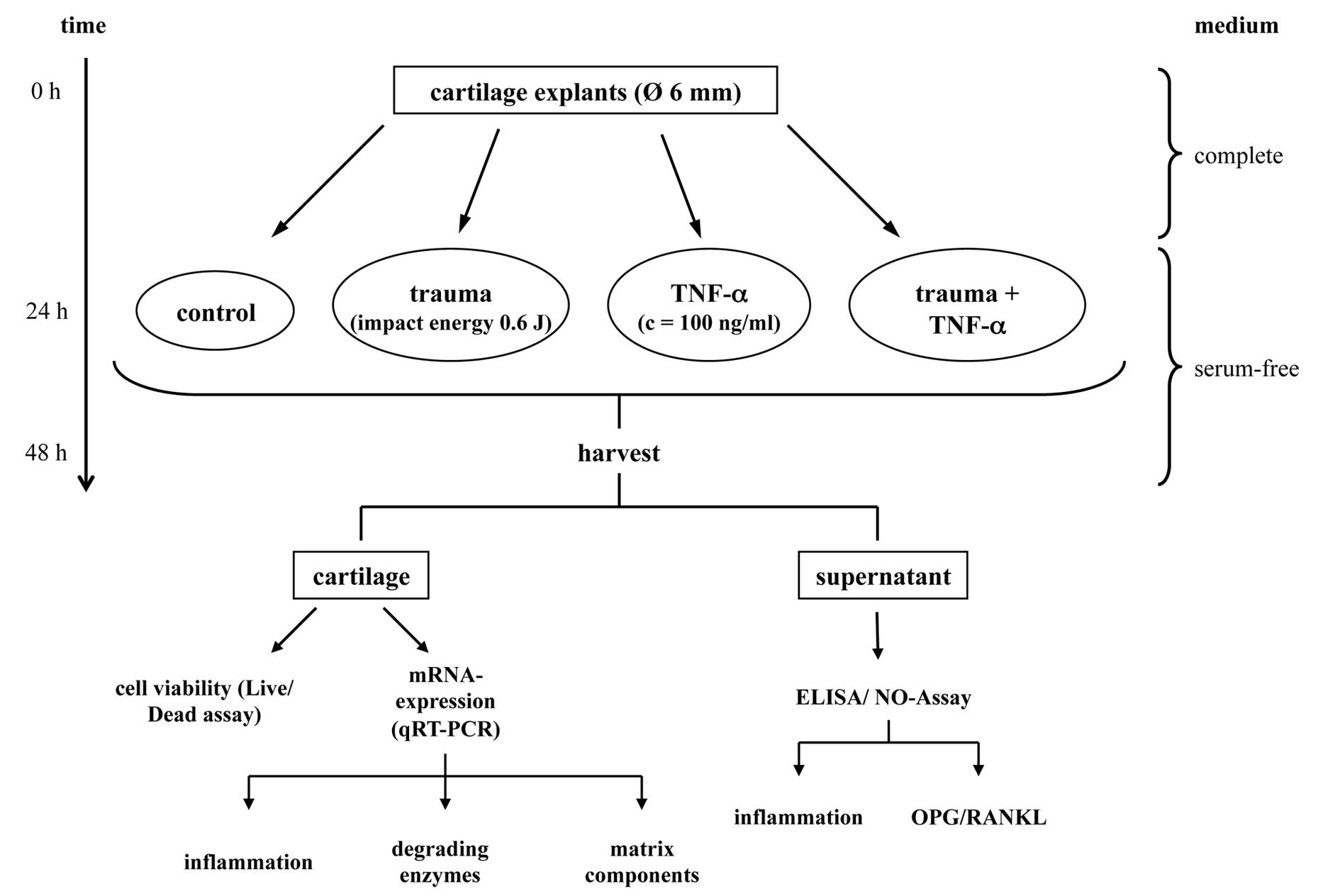
In order to investigate whether TNF-α could alter
the described trauma-induced processes under in vitro
circumstances (Fig. 1) we used
human cartilage, obtained from donors undergoing total knee joint
replacement due to osteoarthritis with informed consent of the
patients according to the terms of the Ethics Committee of the
University of Ulm. Overall, tissue samples from 16 patients were
included in the study. The mean age of the donors was 68±8.1 years
(range 50–78 years). Only tissue from less affected femoral
condyles was used that had a macroscopically smooth surface and no
severe osteoarthritic changes.
Cartilage explants
Full-thickness cartilage explants, 6 mm in diameter,
were harvested by punching the cartilage with a biopsy punch and
separating the cartilage from the underlying subchondral bone with
a scalpel. Each explant was weighed and cultivated in a medium
consisting of 1:1 DMEM/Hams F12 supplemented with 10% fetal bovine
serum, 0.5% penicillin/streptomycin (PAA Laboratories, Pasching,
Austria), 0.5% L-glutamine and 10 μg /ml 2-phospho-L-ascorbic acid
trisodium salt (Sigma-Aldrich, Fluka, Seelze, Germany) for 24 h in
an incubator (37°C, 21% O2, 5% CO2).
Afterwards, the explants were cultivated for 24 h in serum-free
medium consisting of DMEM supplemented with 1% sodium pyruvate,
0.5% L-glutamine, 1% non-essential amino acids, 0.5%
penicillin/streptomycin and 0.1% serum effective substitute (SES-1
solution A). SES-1 solution B was freshly added to the medium in
each case. All chemicals were purchased from Biochrom (Berlin,
Germany) unless specified otherwise.
Impact loading and TNF-α treatment
Blunt injury of the cartilage explants was achieved
with the help of a specially designed drop-tower. It imparts loads
to an indenter, a flat-faced steel rod of 15 mm in diameter,
resting on the explant surface as described previously (19). The impact energy was applied by
dropping a 600 g mass from a height of 10 cm on the indenter, which
resulted in an impact energy of 0.59 J. Unloaded explants served as
controls. In addition to the impact loading, some of the cartilage
explants were treated with TNF-α (100 ng/ml) (PeproTech, Hamburg,
Germany). Twenty-four hours after these treatments the cartilage
explants and the culture media were harvested.
mRNA isolation and cDNA synthesis
For total RNA isolation the cartilage explants were
frozen in liquid N2. After pulverization with a
microdismembrator (Sartorius BBI Systems, Melsungen, Germany), RNA
was isolated using the Lipid Tissue Mini kit (Qiagen, Hilden,
Germany). RNA was reverse transcribed with the Omniscript RT kit
(Qiagen) and used for quantitative real-time PCR-analysis
(StepOnePlus™ Real-Time PCR System, Applied Biosystems, Darmstadt,
Germany).
Real-time polymerase chain reaction
(PCR)
Relative gene expression analysis (2−ΔΔCt
method) using the Platinum®SYBR® qPCR SuperMix UDG
(Invitrogen, Darmstadt, Germany) was used for NOS2A,
5′-ATTCACTCAGCTGTGCATCG-3′ (forward) and 5′-TCAGGTGGGATTTCGAAGAG-3′
(reverse); for COX2, 5′-CCCTTGGGTGTCAAAGGTAA-3′ (forward) and
5′-GGCA AAGAATGCAAACATCA-3′ (reverse); for PTGES, 5′-CCCCC
AGTATTGCAGGAG-3′ (forward) and 5′-GGAAGACCAG GAAGTGCATC-3′
(reverse); for MMP1, 5′-TTCGGGGA GAAGTGATGTTC-3′ (forward) and
5′-ATCTCTGTCGGCAA ATTCGT-3′ (reverse); for COL2A1, 5′-AATGGTGGCTTCC
ATTTCAG-3′ (forward) and 5′-CTGCTTCGTCCAGATAGG CAA (reverse).
TaqMan® Gene Expression Master Mix (Applied Biosystems)
was used for TaqMan® Gene Expression Assay (Applied
Biosystems) Hs00168748_m1 (PTGDS), Hs00968305_m1 (MMP3),
Hs00192708_m1 (ADAMTS4), Hs00199841_m1 (ADAMTS5) and Hs00153936_m1
(ACAN). Power SYBR® Green PCR Master Mix (Applied
Biosystems) was used for 18Sr RNA, 5′-CGC AGCTAGGAATAATGGAATAGG-3′
(forward) and 5′-CAT GGCCTCAGTTCCGAAA-3′ (reverse), which served as
endogenous control. mRNA-expression was determined by real-time PCR
after 24 h, expression levels were normalized to 18Sr RNA.
ELISAs and NO assay
Absolute concentrations of nitrite, a stable
end-product of the NO metabolism, were determined in the media of
the tissue culture using the Griess assay (Griess Reagent System,
Promega, Mannheim, Germany) according to the manufacturer’s
instructions. PGE2 production was measured in the media by PGE2
ELISA Correlate EIA™ kit (Biotrend, Assay Designs, Cologne,
Germany), PGD2 production was measured by Prostaglandin D2 EIA kit
(Cayman, Biomol, Hamburg, Germany) and IL-6, IL-1β and TNF-α
production was measured by Human IL-6 Quantikine ELISA kit, Human
IL-1β/IL-1F2 Quantikine ELISA kit and Human TNF-α Quantikine ELISA
kit, respectively (all R&D Systems, Wiesbaden, Germany),
according to the manufacturer’s instructions. OPG production was
measured by Human Osteoprotegerin/TNFRSF11B DuoSet (R&D
Systems) and RANKL production was measured by ampli-sRANKL ELISA
(Biomedica, Vienna, Austria).
Live/dead cell cytotoxity assay
A Live/Dead® Viability/Cytotoxity assay
(Molecular Probes, Invitrogen, Darmstadt, Germany) was performed to
determine the percentage of viable cells, which was carried out by
staining the chondrocytes of an unfixed tissue section (0.5 mm
thickness) with 1 μM calcein AM and 2 μM ethidium homodimer-1 for
30 min. After washing in PBS, the tissue sections were
microscopically analyzed with the help of a z-stack module
(software AxioVision, Carl Zeiss, Jena, Germany). A quadruplicate
analysis of the basal percentage of living cells showed a standard
deviation of 2.9% and documented good reliability.
Statistical analysis
For standardization of the gene expression levels
determined by real-time PCR-analysis, mRNA-expression was
normalized to 18Sr RNA-expression. Differential regulation was
determined by calculating the treatment-control ratios of gene
expression. The data are presented as box plots, where each box
represents the interquartile range. The line inside the box
displays the median. The whiskers show the minimum and maximum
values. The dependent variables (cell viability, relative gene
expression levels and mediator release) were analyzed by one-way
ANOVA with Bonferroni’s post test for selected pairs of columns
using GraphPad Prism version 5.0 for Mac OS X, GraphPad Software,
San Diego CA, USA, to evaluate significant differences between
different treatment groups. The groups treated with trauma, TNF-α
or trauma plus TNF-α (independent variables) were compared to the
control group. p<0.05 was regarded as significant and p<0.001
as highly significant.
Results
Impacted explants were cultivated with or without
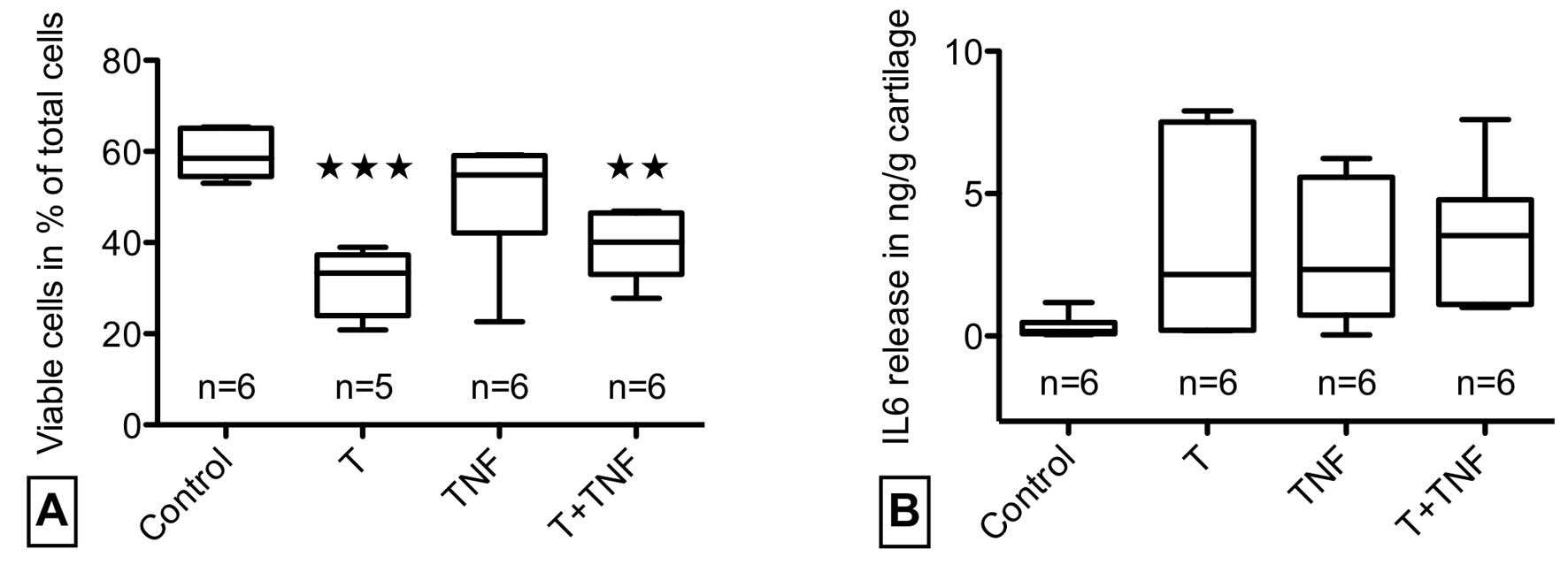
TNF-α for 24 h and processed for Live/dead-staining (Fig. 2A). Quantitative analysis revealed
a significant reduction of vital cells from 59 to 31% after trauma
but no further enhancement of trauma-induced cell death by TNF-α.
TNF-α stimulation of unimpacted explants did not reduce cell
viability.
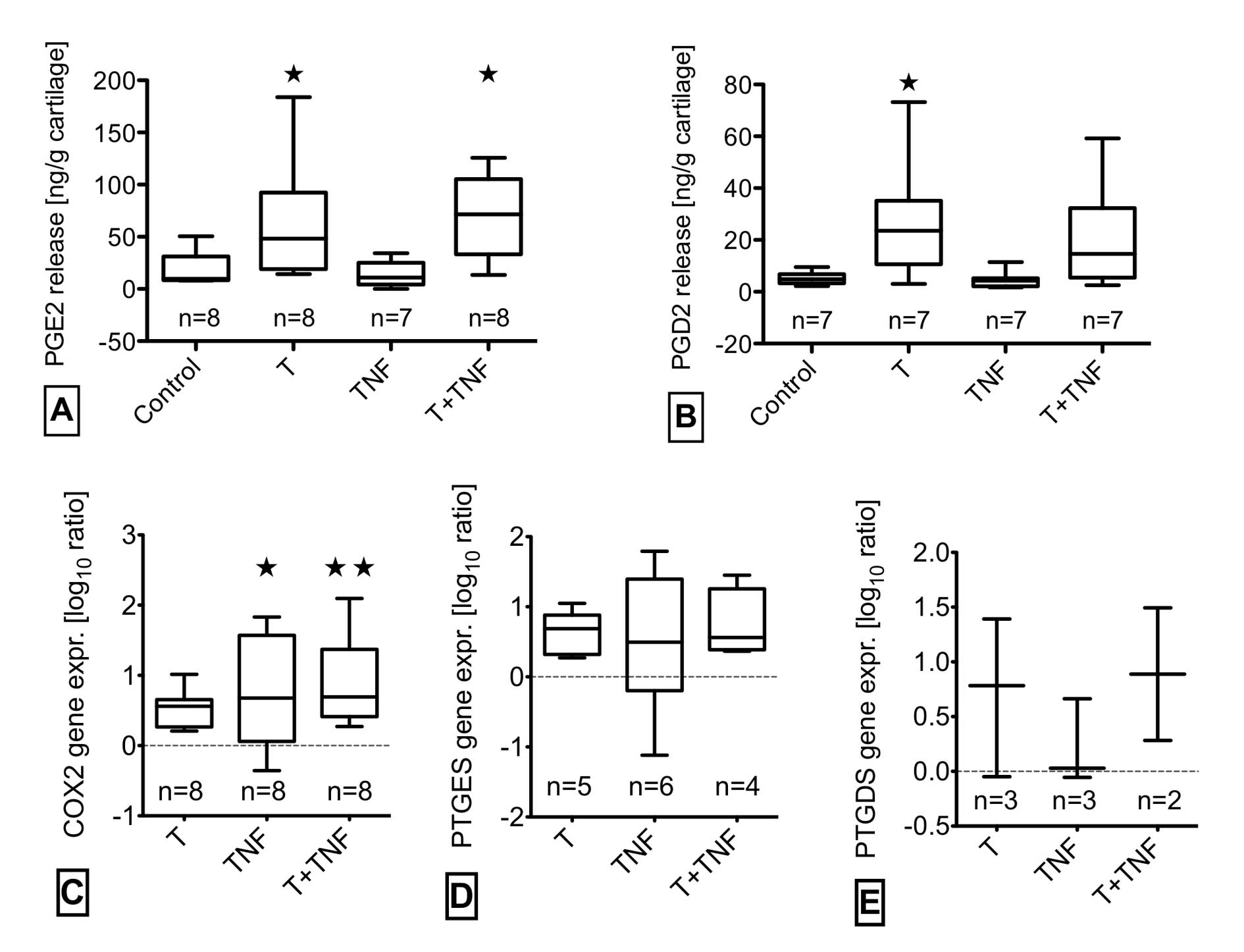
Quantitative analysis of the prostaglandin synthesis
pathway revealed significant effects of trauma with TNF-α
stimulation as shown in Fig. 3.
PGE2 release was significantly increased by trauma with/without
TNF-α whereas TNF-α alone caused no elevation (Fig. 3A). PGE2 synthesis only partly
correlated to the gene expression of the corresponding enzymes at
24 h. We observed a significantly enhanced gene expression of COX2
in TNF-α-stimulated cartilage with/without impact. Trauma alone had
a tendency to elevate COX2 mRNA level (Fig. 3C). PTGES tended to be slightly
increased in trauma with/without TNF-α (Fig. 3D). The production of PGD2 was
elevated significantly by trauma without and in trend by trauma
with TNF-α in correlation with the gene expression of PTGDS
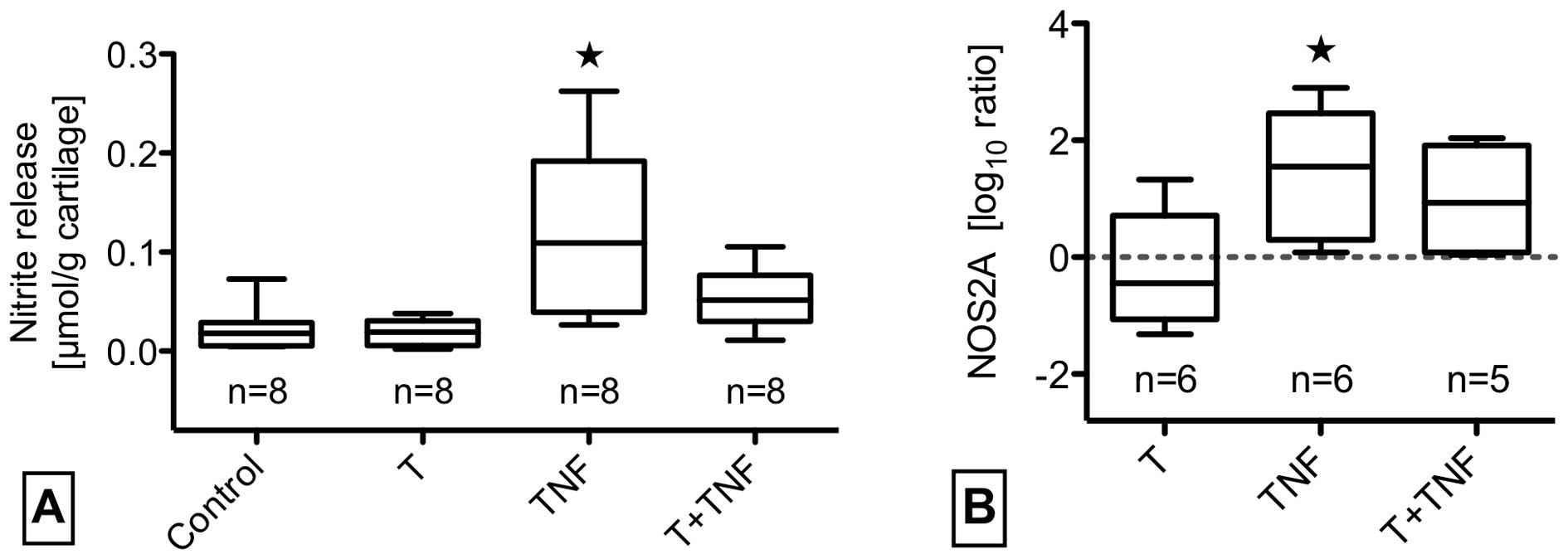
(Fig. 3B and E). The effects of
impact with/without TNF-α on the NO synthesis pathway are shown in
Fig. 4 and revealed a
significantly enhanced NOS2A gene expression and nitrite release by
TNF-α stimulation. Though trauma with TNF-α tended to increase
NOS2A gene expression 30-fold by mean, NO release was hardly
elevated. Using enzyme-linked immunosorbent assay, we determined
the release of IL-6 after trauma and/or TNF-α stimulation by the
cartilage explants. Fig. 2B shows
an elevation of IL-6 release in impacted and/or TNF-α stimulated
explants. A parallel analysis of IL-1β and TNF-α release showed no
activating effect of trauma and TNF-α stimulation caused no
increased IL-1β production (n=9 for control and trauma specimens,
n=3 for TNF-α and TNF-α + trauma specimens, data not shown).
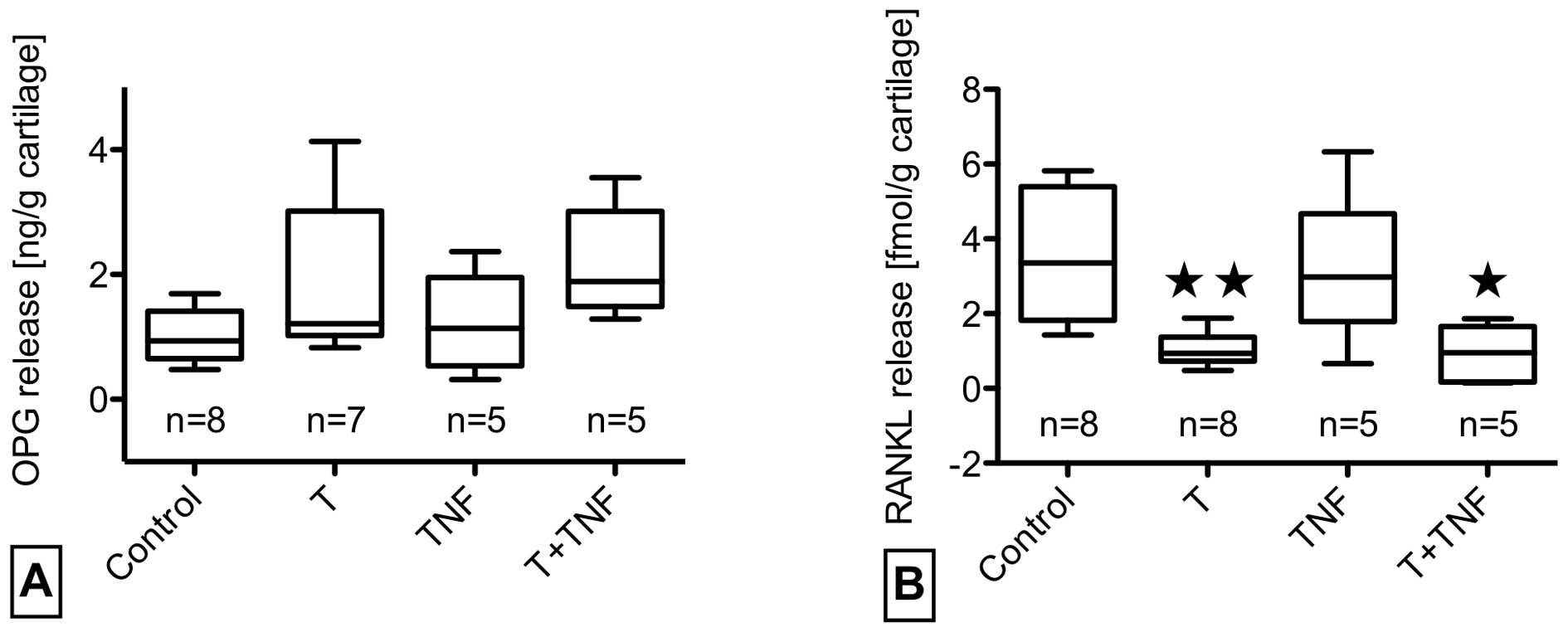
While trauma increased osteoprotegerin release in
half of the cases, concomitant TNF-α stimulation caused a
consistent but not significant elevation. RANKL levels were
significantly reduced by trauma with and without TNF-α. TNF-α
stimulation alone had no influence on OPG/RANKL release (Fig. 5A and B).
Table I shows
trauma and TNF-α effects on gene expression of different degrading
enzymes and matrix components. TNF-α with/without trauma
significantly induced MMP1 gene expression. The combined treatment
resulted in a significantly reduced COL2A1 gene expression and a
trend for lower aggrecan gene expression. The effect on the
expression of these major matrix components of cartilage could be
mainly attributed to the mechanical trauma.
 | Table I.Effects of trauma and TNF-α
stimulation on gene expression of different degrading enzymes and
matrix components. |
Table I.
Effects of trauma and TNF-α
stimulation on gene expression of different degrading enzymes and
matrix components.
| Gene expression
(log10 ratio) relative to control |
|---|
| MMP1 | MMP3 | ADAMTS4 | ADAMTS5 | COL2A1 | ACAN |
|---|
| T | 0.2 (−0.7 to
1.5) | 0.2 (−0.2 to
0.8) | 0.3 (−0.5 to
0.3) | 0.2 (−0.4 to
1.4) | −0.5 (−0.8 to
0.2) | −0.4 (−0.5 to
−0.2) |
| TNF | 0.7a (0.7 to 1.6) | 0.3 (0.1 to
0.5) | −0.4 (−0.9 to
0.1) | −0.3 (−0.5 to
0.2) | −0.2 (−0.5 to
0.1) | −0.1 (−0.6 to
0.4) |
| T + TNF | 0.8a (−0.2 to 1.9) | 0.3 (−0.2 to
1.0) | 0.1 (−0.1 to
0.4) | −0.1 (−1.0 to
0.5) | −0.6a (−1.1 to −0.1) | −0.5 (−1.2 to
0.3) |
Discussion
In synovial joints trauma-induced release of
proinflammatory mediators like TNF-α drive the pathogenetic process
of cartilage degeneration as TNF-α potentiates trauma-induced loss
of proteoglycans (18). We
therefore asked for further synergistic effects and investigated
the single and combined effect of a defined blunt cartilage trauma
and TNF-α exposure on chondrocyte viability, prostaglandin, nitric
oxide, IL-6 and OPG/RANKL metabolism as well as gene expression of
catabolic and anabolic genes in human early-stage osteoarthritic
cartilage explants. The focus on the first 24 h after trauma
disregards possible effects occurring at later time points but
previous studies identified this time frame as determining for cell
survival after cartilage trauma (19) and sufficient to assess possible
apoptotic effects of TNF-α in chondrocytes (21,26).
Twenty-four hours after trauma, a significant
reduction of viable cells to about 50% of untreated samples was
observed, which is comparable to other human cartilage trauma
models (27,28). As trauma did not induce intrinsic
TNF-α release in our in vitro model, the cytokine was only
present after exogenous application and cannot be responsible for
the primary trauma-induced cell death. Even additional stimulation
with exogenous TNF-α at a concentration known to induce apoptosis
under permissive conditions (26)
did not enhance trauma-induced cell death in tissue culture after
24 h. Indeed, TNF-α has previously been reported not to be
proapoptotic in chondrocytes unless they were specifically
sensitized for apoptosis (20,21,26). Possibly, the embedding in the
extracellular matrix may be an essential survival factor for
chondrocytes in tissue culture preventing TNF-α-induced DNA
cleavage (29). As shown here a
preceding single impact trauma known to damage the matrix
mechanically and to cause apoptosis in human cartilage itself
(7), did not induce sensitivity
to TNF-α-mediated apoptosis. One might speculate that the
trauma-associated increase in OPG-expression-although not
statistically significant-may have prevented a further increase in
chondrocyte death by inhibition of TRAIL (TNF-related apoptosis
inducing ligand)-mediated effects as previously described for
myeloma cells (30) since
TRAIL-mediated apoptosis has also been observed in chondrocytes
(31). Anyhow, the results
indicate that stress signaling, matrix deformation and its partial
disruption induced by a single impact trauma does not promote early
TNF-α-mediated apoptosis in human cartilage.
PGE2 and PGD2 release and related gene expression
was increased by trauma, confirming previous studies (19,27). However, additional TNF-α
stimulation did not enhance these trauma-induced effects. Nor did
TNF-α stimulate PGE2 and PGD2 release after 24 h in unimpacted
cartilage, although the mean COX2 gene expression was elevated at
this time point. May be a late induction of gene expression by
TNF-α is not yet reflected in an enhanced PGE2 or PGD2 synthesis
after 24 h. Another reason may arise from possible interactions
between NO and PGE2, since TNF-α lead to highly induced NO levels.
Among versatile effects, NO is described to inhibit PGE2 synthesis
(32,33). NO release could not be induced by
trauma, but was increased by additional TNF-α. Compared to
stimulation of unimpacted cartilage, however, TNF-α induced
NOS2A-expression and NO release was mitigated in case of a
preceeding trauma. This mitigation, again, could be a sign of
interactions between NO and PGE2 metabolism, since trauma induced
high levels of PGE2 (34).
We could show a significant decrease in RANKL and a
trend for an increase in OPG release after trauma. TNF-α could not
enhance this effect. Expression of OPG and RANKL in human cartilage
and chondrocytes has been previously described (25,35). The increased OPG release after
trauma with or without TNF-α could be a consequence of the increase
in PGE2 (36). The decrease of
RANKL after trauma with or without TNF-α indicates the involvement
of another regulatory mechanism related to the mechanical impact.
In fibrochondrocytes of the meniscus it could be shown that cyclic
tensile strain downregulates the expression of RANKL in response to
IL-1β (37). In human
osteoblasts, however, intermittent cyclic tensile strength
increased RANKL expression while OPG-expression was not affected
(38). This indicates a cell-type
specific response or interference of pro-inflammatory stimuli
within the mesenchymal lineage. In the pathophysiologic context of
a blunt cartilage trauma an increase of OPG and a parallel decrease
of RANKL release could possibly contribute to the development of
subchondral sclerosis.
In agreement with previous reports on synergistic
effects of cartilage trauma and TNF-α with respect to proteoglycan
loss (17,18), we found an early increase of MMP1
gene expression after traumatization with TNF-α exposure. MMP1 may
contribute to proteoglycan loss by degradation of the collagenous
network, link protein and aggrecan (39). The reduction of COL2A1 gene
expression by trauma and TNF-α may additionally support the overall
matrix-catabolic effect. The expression of ADAMTS4 and ADAMTS5 was
not affected 24 h after treatment. An induction of these
aggrecanases probably occurs somewhat later since respective
aggrecan cleavage products have been identified in synovial fluid
in the first two weeks after trauma (40). IL-6, which was induced by trauma
and/or TNF-α stimulation in our study, has previously been shown to
enhance proteoglycan loss induced by trauma and TNF-α (18).
The use of well preserved human cartilage from
patients with osteoarthritis in the present study may have
influenced the results because of effects on the basal expression
levels of the genes or mediators studied. The susceptibility of
osteoarthritic chondrocytes to TNF-α should not be diminished, as a
higher number of the p55 TNF-α receptor is expressed in OA
cartilage (14). The advanced age
of the donors means no limitation for this study as the risk for
the development of posttraumatic osteoarthritis even increases with
patient age (41). Furthermore,
the individual responsiveness to proinflammatory stimuli might vary
considerably due to genetical divergence as observed in the context
of osteoarthritis or rheumatoid arthritis (42,43). This fact may also partially
explain the high interindividual variation observed. Nevertheless,
the in vitro system used in this study represents a human
model that allows to analyze cell-biologic effects of defined blunt
cartilage injury in the native 3-dimensional tissue context. Since
several well-known responses to a single impact trauma and to TNF-α
stimulation are preserved it can be regarded as a useful
alternative to similar experimental models with bovine tissue
(17).
In conclusion, we found that exposure of traumatized
human cartilage to TNF-α does not lead to increased chondrocyte
death within the first 24 h. PGE2, PGD2 and IL-6 synthesis was not
markedly modulated by TNF-α while TNF-α-induced NO release was
reduced in case of trauma. In agreement with previous results the
expression of MMP1 was enhanced (15) but expression of ADAMTS4 and
ADAMTS5 was not affected in the early post-traumatic phase. These
results indicate that in the initial phase after blunt cartilage
injury, TNF-α does not potentiate cell death and the release of
inflammatory mediators while degradative processes are rapidly
enhanced (18). Therefore, TNF-α
inhibitors such as etanercept, infliximab or adalimumab, recently
suggested for treatment of joint injuries (5) could be complemented by therapeutics
that mitigate cell death-like anti-oxidants in order to limit
subsequent cartilage damage (44,45). A more detailed knowledge on the
complex interactive mechanisms and signaling pathways in the
initial phase after cartilage trauma may finally lead to bi- or
multidirectional pharmaco-therapeutic approaches to ameliorate
long-term consequences of joint injuries. One promising target in
this context may be the MAPKs that are involved in cell survival,
inflammation and degradative processes (44,46,47).
Acknowledgements
We would like to thank Brunhilde Amann
for excellent technical assistance. This study was supported by the
German Research Council (DFG, grant KFO 200, BR 919/5-1).
References
|
1.
|
M SzczodryCH CoyleSJ KramerP SmolinskiCR
ChuProgressive chondrocyte death after impact injury indicates a
need for chondroprotective therapyAm J Sports
Med3723182322200910.1177/036354650934884019864505
|
|
2.
|
M MajewskiS HabeltK SteinbrückEpidemiology
of athletic knee injuries: A 10-year
studyKnee13184188200616603363
|
|
3.
|
P BakerI ReadingC CooperD CoggonKnee
disorders in the general population and their relation to
occupationOccup Environ
Med60794797200310.1136/oem.60.10.79414504371
|
|
4.
|
M WestinM AlricssonS WernerInjury profile
of competitive alpine skiers: a five-year cohort studyKnee Surg
Sports Traumatol Arthrosc2011751181201222349602
|
|
5.
|
JT LawrenceJ BirminghamAP TothEmerging
ideas: prevention of posttraumatic arthritis through interleukin-1
and tumor necrosis factor-alpha inhibitionClin Orthop Relat
Res46935223526201110.1007/s11999-010-1699-4
|
|
6.
|
DM PhillipsRC HautThe use of a non-ionic
surfactant (P188) to save chondrocytes from necrosis following
impact loading of chondral explantsJ Orthop
Res2211351142200410.1016/j.orthres.2004.02.00215304290
|
|
7.
|
DD D’LimaS HashimotoPC ChenCW ColwellMK
LotzHuman chondrocyte apoptosis in response to mechanical
injuryOsteoarthritis Cartilage9712719200124939082
|
|
8.
|
MA DiMiccoP PatwariPN SiparskyMechanisms
and kinetics of glycosaminoglycan release following in vitro
cartilage injuryArthritis
Rheum50840848200410.1002/art.2010115022326
|
|
9.
|
A AroenS LokenS HeirArticular cartilage
lesions in 993 consecutive knee arthroscopiesAm J Sports
Med32211215200410.1177/036354650325934514754746
|
|
10.
|
Z TangL YangY WangContributions of
different intraarticular tissues to the acute phase elevation of
synovial fluid MMP-2 following rat ACL ruptureJ Orthop
Res27243248200910.1002/jor.2076318846548
|
|
11.
|
K IrieE UchiyamaH IwasoIntraarticular
inflammatory cytokines in acute anterior cruciate ligament injured
kneeKnee109396200310.1016/S0968-0160(02)00083-212649034
|
|
12.
|
MB GoldringKB MarcuCartilage homeostasis
in health and rheumatic diseasesArthritis Res
Ther11224200910.1186/ar259219519926
|
|
13.
|
F De CeuninckL DassencourtP AnractThe
inflammatory side of human chondrocytes unveiled by antibody
microarraysBiochem Biophys Res Commun323960969200415381094
|
|
14.
|
JC FernandesJ Martel-PelletierJP
PelletierThe role of cytokines in osteoarthritis
pathophysiologyBiorheology39237246200212082286
|
|
15.
|
AL StevensJS WishnokFM WhiteAJ
GrodzinskySR TannenbaumMechanical injury and cytokines cause loss
of cartilage integrity and upregulate proteins associated with
catabolism, immunity, inflammation, and repairMol Cell
Proteomics814751489200910.1074/mcp.M800181-MCP20019196708
|
|
16.
|
K DemircanS HirohataK NishidaADAMTS-9 is
synergistically induced by interleukin-1beta and tumor necrosis
factor alpha in OUMS-27 chondrosarcoma cells and in human
chondrocytesArthritis
Rheum5214511460200510.1002/art.2101015880812
|
|
17.
|
P PatwariMN CookMA DiMiccoProteoglycan
degradation after injurious compression of bovine and human
articular cartilage in vitro: interaction with exogenous
cytokinesArthritis Rheum4812921301200310.1002/art.10892
|
|
18.
|
Y SuiJH LeeMA DiMiccoMechanical injury
potentiates proteoglycan catabolism induced by interleukin-6 with
soluble interleukin-6 receptor and tumor necrosis factor alpha in
immature bovine and adult human articular cartilageArthritis
Rheum6029852996200910.1002/art.24857
|
|
19.
|
H JoosC HogrefeL RiegerL DurselenA
IgnatiusRE BrennerSingle impact trauma in human early-stage
osteoarthritic cartilage: Implication of prostaglandin D2 but no
additive effect of IL-1beta on cell survivalInt J Mol
Med282712772011
|
|
20.
|
H KimWS SongTNF-alpha-mediated apoptosis
in chondrocytes sensitized by MG132 or actinomycin DBiochem Biophys
Res Commun295937944200210.1016/S0006-291X(02)00789-112127985
|
|
21.
|
MJ López-ArmadaB CaramésM
Lires-DeánCytokines, tumor necrosis factor-alpha and
interleukin-1beta, differentially regulate apoptosis in
osteoarthritis cultured human chondrocytesOsteoarthritis
Cartilage14660669200616492401
|
|
22.
|
MB GoldringSR GoldringOsteoarthritisJ Cell
Physiol213626634200710.1002/jcp.2125817786965
|
|
23.
|
D UmlaufS FrankT PapJ BertrandCartilage
biology, pathology, and repairCell Mol Life
Sci6741974211201010.1007/s00018-010-0498-020734104
|
|
24.
|
AE KearnsS KhoslaPJ KostenuikReceptor
activator of nuclear factor kappaB ligand and osteoprotegerin
regulation of bone remodeling in health and diseaseEndocr
Rev29155192200810.1210/er.2007-001418057140
|
|
25.
|
H KomuroT OleeK KühnThe
osteoprotegerin/receptor activator of nuclear factor
kappaB/receptor activator of nuclear factor kappaB ligand system in
cartilageArthritis
Rheum4427682776200110.1002/1529-0131(200112)44:12%3C2768::AID-ART464%3E3.0.CO;2-I11762937
|
|
26.
|
F YoshimuraH KannoM UzukiK TajimaT
ShimamuraT SawaiDownregulation of inhibitor of apoptosis proteins
in apoptotic human chondrocytes treated with tumor necrosis
factor-alpha and actinomycin DOsteoarthritis
Cartilage14435441200610.1016/j.joca.2005.11.00316368252
|
|
27.
|
JE JeffreyRM AspdenCyclooxygenase
inhibition lowers prostaglandin E2 release from articular cartilage
and reduces apoptosis but not proteoglycan degradation following an
impact load in vitroArthritis Res Ther9R129200710.1186/ar2346
|
|
28.
|
DD D’LimaS HashimotoPC ChenCW ColwellMK
LotzImpact of mechanical trauma on matrix and cellsClin Orthop
Relat ResSuppl 391S90S99200111603728
|
|
29.
|
BA FischerS MundleAA ColeTumor necrosis
factor-alpha induced DNA cleavage in human articular chondrocytes
may involve multiple endonucleolytic activities during
apoptosisMicrosc Res
Tech50236242200010.1002/1097-0029(20000801)50:3%3C236::AID-JEMT7%3E3.0.CO;2-E
|
|
30.
|
CM ShipmanPI CroucherOsteoprotegerin is a
soluble decoy receptor for tumor necrosis factor-related
apoptosis-inducing ligand/Apo2 ligand and can function as a
paracrine survival factor for human myeloma cellsCancer
Res639129162003
|
|
31.
|
I PettersenY FigenschauE OlsenW BakkelundB
SmedsrodB SveinbjornssonTumor necrosis factor-related
apoptosis-inducing ligand induces apoptosis in human articular
chondrocytes in vitroBiochem Biophys Res
Commun296671676200210.1016/S0006-291X(02)00916-6
|
|
32.
|
AR AminM AtturSB AbramsonNitric oxide
synthase and cyclooxygenases: distribution, regulation, and
intervention in arthritisCurr Opin
Rheumatol11202209199910.1097/00002281-199905000-0000910328580
|
|
33.
|
F GuilakB FermorFJ KeefeThe role of
biomechanics and inflammation in cartilage injury and repairClin
Orthop Relat
Res4231726200410.1097/01.blo.0000131233.83640.9115232421
|
|
34.
|
P MarottaL SautebinM Di RosaModulation of
the induction of nitric oxide synthase by eicosanoids in the murine
macrophage cell line J774Br J
Pharmacol107640641199210.1111/j.1476-5381.1992.tb14499.x1282071
|
|
35.
|
S Kwan TatN AmiableJP PelletierModulation
of OPG, RANK and RANKL by human chondrocytes and their implication
during osteoarthritisRheumatology
(Oxford)4814821490200919762475
|
|
36.
|
J Moreno-RubioG Herrero-BeaumontL TardioMA
Alvarez-SoriaR LargoNonsteroidal antiinflammatory drugs and
prostaglandin E(2) modulate the synthesis of osteoprotegerin and
RANKL in the cartilage of patients with severe knee
osteoarthritisArthritis
Rheum62478488201010.1002/art.2720420112374
|
|
37.
|
J DeschnerE WypasekM FerrettiB RathM
AnghelinaS AgarwalRegulation of RANKL by biomechanical loading in
fibrochondrocytes of meniscusJ
Biomech3917961803200610.1016/j.jbiomech.2005.05.03416038916
|
|
38.
|
L KrejaA LiedertS HasniL ClaesA
IgnatiusMechanical regulation of osteoclastic genes in human
osteoblastsBiochem Biophys Res
Commun368582587200810.1016/j.bbrc.2008.01.10618243138
|
|
39.
|
MD SternlichtZ WerbHow matrix
metalloproteinases regulate cell behaviorAnnu Rev Cell Dev
Biol17463516200110.1146/annurev.cellbio.17.1.46311687497
|
|
40.
|
A StruglicsM HanssonLS LohmanderHuman
aggrecanase generated synovial fluid fragment levels are elevated
directly after knee injuries due to proteolysis both in the inter
globular and chondroitin sulfate domainsOsteoarthritis
Cartilage1910471057201110.1016/j.joca.2011.05.006
|
|
41.
|
H RoosT AdalberthL DahlbergLS
LohmanderOsteo-arthritis of the knee after injury to the anterior
cruciate ligament or meniscus: the influence of time and
ageOsteoarthritis
Cartilage3261267199510.1016/S1063-4584(05)80017-28689461
|
|
42.
|
EM SchneiderW DuJ FiedlerThe (−765
G->C) promoter variant of the COX-2/PTGS2 gene is associated
with a lower risk for end-stage hip and knee osteoarthritisAnn
Rheum Dis70145814602010
|
|
43.
|
TC Van der Pouw KraanFA van GaalenPV
KasperkovitzRheumatoid arthritis is a heterogeneous disease:
evidence for differences in the activation of the STAT-1 pathway
between rheumatoid tissuesArthritis Rheum4821322145200312905466
|
|
44.
|
L DingE HeyingN NicholsonMechanical impact
induces cartilage degradation via mitogen activated protein
kinasesOsteoarthritis
Cartilage1815091517201010.1016/j.joca.2010.08.01420813194
|
|
45.
|
W GoodwinD McCabeE SauterRotenone prevents
impact-induced chondrocyte deathJ Orthop
Res2810571063201020108345
|
|
46.
|
K TakebeT NishiyamaS HayashiRegulation of
p38 MAPK phosphorylation inhibits chondrocyte apoptosis in response
to heat stress or mechanical stressInt J Mol
Med27329335201121181091
|
|
47.
|
J RadonsAK BosserhoffS GraesselW FalkTEO
Schubertp38MAPK mediates IL-1-induced down-regulation of aggrecan
gene expression in human chondrocytesInt J Mol
Med17661668200616525725
|