Introduction
It is estimated that approximately 180 million
people have been infected with the hepatitis C virus (HCV) and
approximately 130 million people are chronic HCV carriers (1). HCV is a common cause of chronic
hepatitis, cirrhosis and hepatocellular carcinoma worldwide
(2–4). Currently, there are two main methods
for detecting an HCV infection: one detects viral RNA by RT-PCR
(5–8) and the other detects HCV antibodies
by immunoassay [enzyme-linked immunosorbent assay (ELISA)] in serum
(9,10). The sensitive ELISA assay uses
recombinant viral proteins corresponding to multiple polypeptides
from different viral regions, including structural proteins and
non-structural polypeptides (11–13). It can be used as a confirmation
test. However, its usage is limited in the clinical setting as the
procedure requires sophisticated laboratory equipment and there is
a high probability of contamination. Therefore, the purpose of our
study was to develop an affordable and reliable rapid lateral flow
test to detect the presence of HCV antibodies in blood samples by
screening for HCV antigens, which would help decrease the chances
of HCV infection from blood transfusions.
Materials and methods
Plasmids and bacterial strains
The p90/HCVFLlongpU plasmid carrying full-length
coding sequences of HCV was a generous gift from Professor Charles
M. Rice from the Center for the Study of Hepatitis C, Rockefeller
University, New York, NY, USA. The Escherichia coli (E.
coli) strains, Jm109, DH5a and BL21 (DE3), were used as the
cloning and expression hosts.
Reagents and instruments
A panel of 23 standard positive sera, 8 standard
negative sera, a set of quality control references for anti-HCV
detection that contain known amounts of anti-HCV antibodies (Artron
BioResearch Inc., Burnaby, BC, Canada) and 300 clinical sera were
used for the antigenicity assessment of HCV proteins. Other
reagents and instruments included goat anti-mouse HCV IgG
polyclonal antibody, 30–60 nm colloidal gold particles (from Artron
BioResearch Inc.), HCV-ELISA (KHB, Shanghai, China), a
NanoDrop® ND-1000 Spectrophotometer, a Bio-Rad BioLogic
LP, ZQ4000 test strip cutter, and XYZ-3000 Bio-Dot (all from
Bio-Rad, Shanghai, China).
Construction and expression of
recombinant HCV antigens
To obtain the HCV antigens, the sequences encoding
the desired regions in the HCV genome were amplified by RT-PCR, and
cloned into the prokaryotic expression vectors, pQE30 (Qiagen,
Hilden, Germany), pET32a(+) (Novagen, Darmstadt, Germany), or
pGEX-4T-2 (GE Healthcare Life Sciences, Chalfont St. Giles, UK),
in-frame downstream of the 6-His-tag or glutathione S-transferase
(GST)-tag coding sequence. Primers used for the HCV PCR
amplification are listed in Table
I and the structures of the plasmids are illustrated in
Fig. 1. E. coli BL21 (DE3)
cells harbouring the HCV gene fragment were grown at 37°C in LB
medium containing 50 μg/ml of ampicillin to OD600 = 0.8.
The expression of the antigens was induced by adding
isopropyl-β-D-thiogalactopyranoside (IPTG) to a final concentration
of 1 mmol/l. The cells were harvested 4–6 h later by centrifugation
at 10,000 rpm for 15 min and stored at −20°C. Solubility analyses
of expression products were performed as previously described
(14). Briefly, harvested
bacteria were re-suspended in phosphate-buffered saline (PBS;
containing 140 mmol/l NaCl, 2.7 mmol/l KCl, 10 mmol/l
Na2HPO4, 1.8 mmol/l
KH2PO4, pH 7.3), sonicated on an ice-bath,
and centrifuged at 10,000 rpm for 20 min at 4°C. After
centrifugation, the soluble and insoluble fractions were analyzed
for the presence of expression products.
 | Table I.Sequences of oligonucleotide primers
that were used to validate the expression of several different HCV
segment genes. |
Table I.
Sequences of oligonucleotide primers
that were used to validate the expression of several different HCV
segment genes.
| No. | Primer
sequences | Primary
description | Enzyme site |
|---|
| F1 | 5′-CGGGATCCATGAGCACGAATCCTAAACC-3′ | Core-190 | BamHI |
| R1 | 5′-TTAAGCTTCTGAAGCGGGCACAGTC-3′ | | HindIII |
| F2 | 5′-CGGGTACCATGAGCACGAATCCTAAAC-3′ | Core-68 | KpnI |
| R2 | 5′-TTGGATCCACGTGCCTTGGGGATA-3′ | | BamHI |
| F3 | 5′-CGGGTACCACGCAAGACTGCAATTGTT-3′ | E1 300–407 | KpnI |
| R3 | 5′-TTAAGCTTGCTTGGCGCCTGGT-3′ | | HindIII |
| F4 | 5′-CGGGATCCTACCAAGTGCGCAATTC-3′ | E1 192–318 | BamHI |
| R4 | 5′-CGAAGCTTATGCCATGCGATGACC-3′ | | HindIII |
| F5 | 5′-AAGGATCCACACCAGGCGCCAAG-3′ | E2 403–642 | BamHI |
| R5 | 5′-TTGAATTCCGCTTCCAGCCTGTG-3′ | | EcoRI |
| F6 | 5′-CCGGTACCTTGGAGAACCTCGTAAT-3′ | P7 747–807 | KpnI |
| R6 | 5′-TGGAATTCGTATGCCCGCTGAG-3′ | | EcoRI |
| F7 | 5′-TTGGTACCTGTGGCGGCGTTGTT-3′ | NS2 817–1022 | KpnI |
| R7 | 5′-CGGAATTCCCACCCCTTGGAGACCAT-3′ | | EcoRI |
| F8 | 5′-TTGGTACCCAGACGAGAGGCCTCCTAG-3′ | NS3 1034–1193 | KpnI |
| R8 | 5′-TTGGATCCGTCCACCGCCTTAGCC-3′ | | BamHI |
| F9 | 5′-AAGGATCCGTGTGCACCCGTGGAGT-3′ | NS3 1183–1476 | BamHI |
| R9 | 5′-GGAAGCTTGGAGCGTGGTTGTCTCAAT-3′ | | HindIII |
| F10 | 5′-CCGAATTCTTCAGCCTTGACCCTAC-3′ | NS3 1463–1656 | EcoRI |
| R10 | 5′-TTAAGCTTGCGTGACGACCTCC-3′ | | HindIII |
| F11 | 5′-TTGGTACCGTCCTGGCTGCTCTG-3′ | NS4A 1665–1710 | KpnI |
| R11 | 5′-CGAAGCTTGGCACTCTTCCATCTCA-3′ | | HindIII |
| F12 | 5′-TTGGTACCCCTGGAGCCCTTGTAGT-3′ | NS4B 1888–1937 | KpnI |
| R12 | 5′-TTGAATTCGCTCTCCGGCACGTAG-3′ | | EcoRI |
| F13 | 5′-AAGGTACCGCAGAGGAGGATGAGC-3′ | NS5A 2258–2419 | KpnI |
| R13 | 5′-CGGAATTCGCAGCACACGACATCTT-3′ | | EcoRI |
| F14 | 5′-TTGGTACCTGGACAGGCGCACTCGT-3′ | NS5B 2425–2733 | KpnI |
| R14 | 5′-TTGTCGACCGAGCATGGTGCAGTCC-3′ | | SalI |
Purification of recombinant HCV
antigens
To purify the expressed proteins, we chose to use a
Ni-nitrilotriacetic acid (Ni-NTA) affinity chromatography column
for His-tagged proteins and a glutathione sepharose™ 4B column for
GST-tagged proteins. The two methods are similar as regards
experimental procedures but differ in column chromatography and
reagents, as described below:
i) Protein purification with Ni-NTA column.
The column was first equilibrated with lysis buffer (50 mM
NaH2PO4, 300 mM NaCl, 10 mM imidazole, pH
7.8) at five times the volume of the beads. The sample was then
loaded and allowed to flow slowly in order to maximize the amount
of protein bound to the beads. The flow-through solution was
collected for SDS-PAGE analysis later. After the sample was
completely loaded, washing buffer (50 mM
NaH2PO4, 300 mM NaCl, 20 mM imidazole, pH
8.0) was added to wash off unspecific proteins bound to the beads
or remaining in the column until the OD280 reading was
below 0.100. The proteins of interest were eluted by adding elution
buffer (50 mM NaH2PO4, 300 mM NaCl, 250 mM
imidazole, pH 8.0).
ii) Protein purification with glutathione
sepharose 4B column. The general procedures were the same as
those for the Ni-NTA columns, although the buffers used in the GST
columns were different. PBS solution (pH 7.4) was used for
equilibration. After the sample was loaded, the same PBS solution
was used to wash the column. Finally, the glutathione solution (50
mM Tris-HCL, 10 mM reduced glutathione) was used to elute the
protein of interest. All purified recombinant HCV antigens were
found to be >90% pure based on SDS/polyacrylamide gel analysis
followed by Coomassie blue staining. Eluents with high
OD280 were collected into a membrane bag and dialysis
was performed overnight at 4°C in a buffer (PBS buffer). Protein
concentration was measured using the Bradford method with bovine
serum albumin as the standard.
Construction of double antigen
sandwich-lateral flow immunoassay (DAS-LFIA) strip and indirect
lateral flow immunoassay (I-LFIA)
The DAS-LFIA device for the detection of anti-HCV
antibodies was manufactured by Artron BioResearch Inc. First, we
optimized the conditions for the colloidal gold conjugation of the
purified antigen and the coating of the gold-conjugated recombinant
protein on non-woven fabric sheets. Colloidal gold is used as an
indicator for the presence of antigens binding to the membrane on
the rapid lateral flow test strip. After the proteins are bound to
colloidal gold to form the conjugate, the fabric sheets are placed
in a dry room for at least 2 h for drying. Subsequently, the
purified antigen, diluted in PBS, is coated on the test region.
Simultaneously, HCV IgG polyclonal antibody, diluted in PBS, is
coated on the control region. The coated membrane is dried for a
minimum of 24 h and then blocked with a particular blocking
solution. Finally, the test strip is assembled such that everything
slightly overlaps in order to allow for the continuous lateral flow
of the liquid sample. The test strip has an absorption pad, a strip
of membrane, a conjugate fibre and other fibres to hold the
conjugate fibre in place. Instructions on how to assemble a test
strip were provided by Artron BioResearch Inc. The combination of
the coating protein, conjugate protein, conjugate fibre and
dialysis buffer forms a ‘system’ in a test strip. The test strip
developed was tested with different positive and negative HCV sera.
I-LFIA was manufactured using IgG antibody conjugating colloidal
gold instead of antigen.
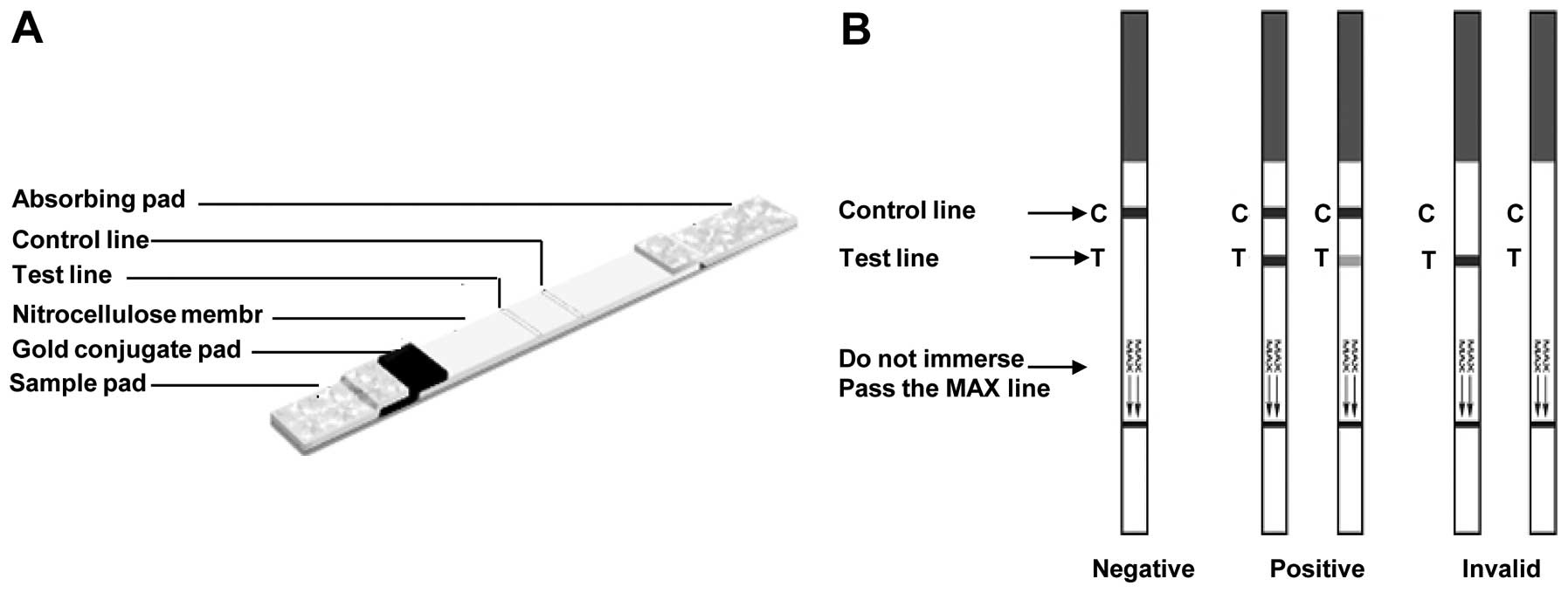
Test principle and assay procedure
Each test solution (50 μl) was pipetted onto the
sample pad and driven to migrate by capillary action along the
strip. If HCV antibodies were present in the serum or plasma, they
would react with the colloidal gold-conjugated antigen to form an
antibody-antigen complex. This complex would flow through the
absorbent device and bind to the antigen in the positive reaction
test zone (‘T’ area), forming a gold-conjugated Ag-Ab-Ag sandwich
complex, producing a pink-purple colored band. A colored band in
the control region of the device indicates adequate sample volume
and capillary action. The absence of a colored band in the control
region is an indication of an invalid result. Positive results were
read as soon as two colored bands appeared. Negative samples
provided only one pink control band. If no control band was
present, the test was considered invalid. Color formation for both
reactions was complete after 5–10 min. A schematic representation
of possible test results is shown in Fig. 2B.
Determination of sensitivity and
specificity of the HCV strip
To assess the sensitivity of the HCV strip, a panel
of 23 standard positive sera were simultaneously measured by the
ELISA, DAS-LFIA, I-LFIA and RT-PCR methods. In addition, serial
dilutions of a reference panel with known amounts of anti-HCV
antibodies with a concentration from 8 NCU/ml (NCU meaning national
clinical unit) to 0.5 NCU/ml were measured by the DAS-LFIA strip.
To assess the specificity and accuracy the ddH20,
positive enhancement sample (a set of quality control references
for anti-HCV antibody detection that contains known amounts of
anti-HCV antibodies; Artron BioResearch Inc.), BS control and 8
HCV-negative patients were analyzed by the DAS-LFIA strip.
Detection of HCV antibodies in clinical
specimens
A total of 300 clinical samples was analyzed by the
new DAS-LFIA method as described above and the HCV ELISA test kit
(Shanghai Huaguan Biochip Co., Ltd.) according to the supplier’s
instructions.
Statistical analysis
The statistical package SPSS 11.5 was used for data
analysis. P-value ≤0.05 was considered to indicate a statistically
significant difference.
Results
Construction of expression plasmids
For the expression of HCV proteins in E.
coli, corresponding coding sequences were cloned into the
histidine fusion expression vectors, pET32a(+) and pQE30, or the
GST-tag expression vector, pGEX-4T-2, as shown in Table I and Fig. 1. Recombinant plasmids were
examined and confirmed by PCR amplification, restriction enzyme
digestion and DNA sequencing. We successfully produced a set of
recombinant proteins derived from HCV structural (core, E1 and E2)
and non-structural proteins (NS2, NS3, NS4A, NS4B, NS5A and NS5B)
in BL21 (DE3) cells by using the E. coli expression
system.
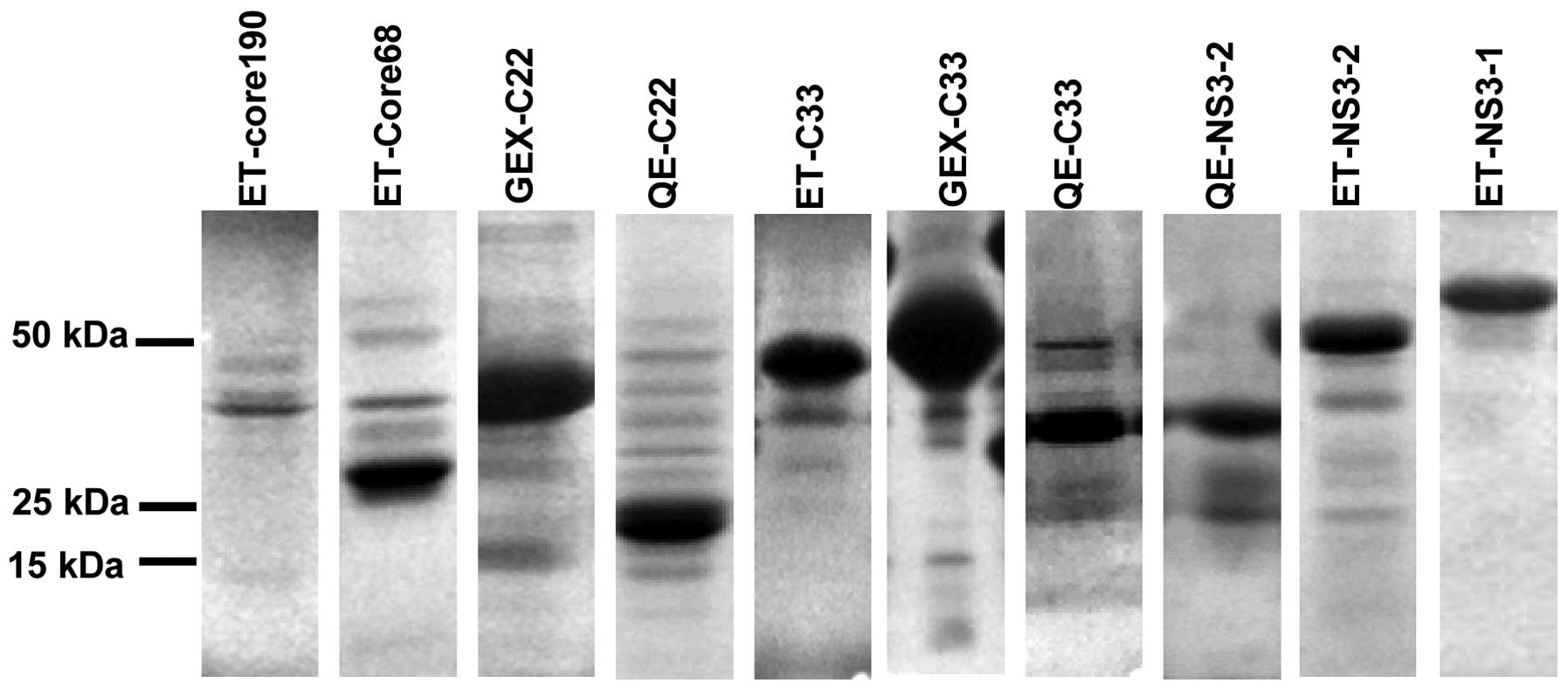
Expression and purification of
recombinant HCV proteins
In order to obtain pure proteins, recombinant
plasmids were used to transform E. coli BL21 (DE3) cells and
expression was induced with 1 mM IPTG. The expressed proteins were
purified with Ni2+-chelate affinity chromatography and a
glutathione sepharose™ 4B column. Proteins were examined using
SDS-PAGE (Fig. 3). The position
of the protein bands was consistent with the expected molecular
weight of the different HCV segments. Protein concentration was
determined by the BCA method, using BSA as the standard. Our
further analyses demonstrated that the HCV proteins were pure and
that they could be used directly for the construction of the HCV
DAS-LFIA strip.
Analytical parameters of the optimized
one-step strip
In the current study, we optimized the concentration
of the coating antigen, the amount of colloidal gold-labelled
antigen on the conjugate pad, the characteristics of the materials
used, the buffer systems, the additives, and the solvents applied
to ensure that the assays ran successfully over the concentration
range required (Table II).
| Table II.Specification of materials and
parameters of the optimized DAS-LFIA strip. |
Table II.
Specification of materials and
parameters of the optimized DAS-LFIA strip.
| Item | Specification |
|---|
| Membrane | High-flow NC
membrane; thickness, 140 μm±20%; absorption: speed, ≥10 mm/min;
size of the pore, 5–15 μm |
| Fiber glass | Absorbent cotton:
thickness, 0.3–0.5 mm; intensity, 50±5 g/m2 |
| Absorbent
paper | Absorbent cotton:
thickness, 0.6–0.8 mm; intensity, 270±20 g/m2 |
| Colloidal gold | Particle size,
30–60 nm |
| Coating buffer | Tris-HCL buffer pH
8.0 |
| Coating
concentration | 1 mg/ml |
Sensitivity and specificity of HCV
DAS-LFIA strip including core and a new NS3 recombinant
protein
The purified proteins would be used to determine
whether they have a prognostic value in patients suffering from
chronic HCV infection. Of the 23 standard positive sera (derived
from confirmed HCV RNA and antibody-positive patients), 95.6%
recognized the core and 95.6% recognized the NS3 protein. The
remaining HCV proteins were very poorly immunogenic. Only three
serum specimens recognized the NS5B protein and none of the sera
recognized the NS2 protein.The results showed that the core and NS3
[1183–1476 amino acids (aa)] of the HCV polyprotein had strong
positive reactions to positive sera. However, the core and NS3
(1183–1476 aa) each had one false-negative result when testing the
positive control samples. Thus, we combined the core and NS3
(1183–1476 aa) as the coating antigen of the new DAS-LFIA strip.
Our data suggested that the new DAS-LFIA strip was able to detect
HCV antibodies at high positive rates (100%) when compared with the
ELISA, I-LFIA and RT-PCR methods using the same serum samples
(Table III) (P>0.05). In this
model of HCV infection, the test line of the anti-HCV DAS-LFIA
strip was 2 NCU/ml (Table IV).
All antigens ran against negative samples did not show any signs of
reactivity in comparison to the control test strip (Fig. 4). This indicated that the HCV
antigen did not cross-react with other common viral antibodies and
thus, was highly specific to HCV.
| Table III.Results of the positive rates of
anti-HCV antibodies detected by the DAS-LFIA strip and the other
methods. |
Table III.
Results of the positive rates of
anti-HCV antibodies detected by the DAS-LFIA strip and the other
methods.
| Tests | Positive rate
(%) |
|---|
| ELISA (KHB) | (22/23) 95.65 |
| DAS-LFIA (core and
NS 1183–1476 aa) | (23/23) 100 |
| I-LFIA (core and NS
1183–1476 aa) | (22/23) 95.65 |
| I-LFIA (core) | (22/23) 95.65 |
| I-LFIA (NS3
1183–1476 aa) | (22/23) 95.65 |
| I-LFIA (NS3
1192–1457 aa) | (20/23) 86.95 |
| RT-PCR | (23/23) 100 |
| Table IV.Lowest test limit for a positive
human anti-HCV antibody detected by the DAS-LFIA strip. |
Table IV.
Lowest test limit for a positive
human anti-HCV antibody detected by the DAS-LFIA strip.
| Human anti-HCV
antibody concentration (NCU/ml) | LFIA result |
|---|
| 8 | Positive |
| 4 | Positive |
| 2 | Positive |
| 1 | Negative |
| 0.5 | Negative |
Immunoassay of the 300 clinical
specimens
A total of 300 samples was measured using the
anti-HCV DAS-LFIA strip and anti-HCV ELISA immunoassays. In these
300 cases, we found that the rate of the anti-HCV DAS-LFIA strip
and the ELISA-negative one was 78% (234/300). The rate of anti-HCV
DAS-LFIA-negative but ELISA-positive was 2% (6/300). The rate of
the anti-HCV DAS-LFIA-positive strip but the ELISA-negative one was
3.67% (11/300). The rate of the anti-HCV DAS-LFIA and
ELISA-positive was 16.33% (49/300). The concordance between the
ELISA and DAS-LFIA methods was 94.33%. The disagreement rate
between the ELISA and DAS-LFIA methods was not significant
(χ2= 0.941, P= 0.332) (Table V).
| Table V.Results from 300 plasma donor samples
detected by the DAS-LFIA strip and HCV ELISA assay. |
Table V.
Results from 300 plasma donor samples
detected by the DAS-LFIA strip and HCV ELISA assay.
| HCV ELISA
|
|---|
| DAS-LFIA | Negative | Positive | Total |
|---|
| Negative | 234 | 6 | 240 |
| Positive | 11 | 49 | 60 |
| Total | 245 | 55 | 300 |
Discussion
In this present study, we characterized the
immunoreactivity of recombinant HCV polypeptides derived from many
different regions of the HCV polyprotein expressed in bacteria. The
purpose of synthesizing these segments is to determine which
segments encoded by the genome are significant for the development
of anti-HCV assays and to find a multiple epitope fusion antigen
which incorporates all of the major immunodominant epitopes from
the functional regions of the HCV genome. Therefore, we first
constructed the different HCV segments into vectors containing
His-tag or GST-tag to induce expression. The proteins were
expressed in BL21 (DE3) cells as fusion proteins with a 26 kDa
GST-tag or 18 kDa His-tag used for detection and affinity
purification. The immunogenicity of the tagged fusion proteins was
analyzed using HCV strip analysis with standard positive and
negative anti-HCV sera. Our results showed that the recombinant
soluble proteins were expressed successfully. The sera studied
recognized the core and NS3 protein at very high levels, whereas
the other proteins, such as NS4B, NS4A, NS5A and NS5B, were found
to have lower levels of reactivity. The E2 protein rarely reacted
with anti-HCV positive serum. Thus, the core protein in combination
with a new NS3 protein (1183–1476 aa) constitutes almost all of the
major immunogenic proteins of the HCV. However, this study does not
exclude the possibility that the low reactivity to some HCV
antigens is due to a quantitative reduction in the titer of
antibodies and not due to an absence of reactivity.
To determine the contributions of various regions of
the core protein and NS3 protein to infectivity diagnosis, clones
of the core gene and different NS3 segments were expressed in E.
coli cells and the recombinant proteins were used to test human
anti-HCV-positive sera in the ELISA and the rapid lateral flow test
strip. The data suggested that the full-length core protein and the
new NS3 protein (1183–1476 aa) were suitable for analyzing the
presence of antibodies against individual HCV proteins in human
sera obtained from patients suffering from chronic HCV infection.
These results are in agreement with those of previous studies in
which the putative nucleocapsid protein (C) and non-structural
proteins (NS3) were found to contain the most immunodominant
epitopes (15–19).
Several studies have indicated that a peptide
spanning aa 2–120 of the C gene (C22) is a major component of the
commercially available second-generation anti-HCV tests. Our study
demonstrates that the full-length core protein has better
reactivity than C22. It is possible that the C-terminal domain of
the core protein contains key peptide sequences for constructing
the viral particle and regulating viral assembly (4,20).
The HCV NS3 protein is composed of an amino terminal
protease and a carboxyl terminal RNA helicase (21). NS3 contains major antigenic
epitopes and plays an important role in the diagnosis of HCV
infection, particularly in early HCV infection (21). In this study, to our knowledge,
our results demonstrate for the first time that a new NS3 segment
(1183–1476 aa) shows strong reactivity in 95.6% of RT-PCR-positive
samples and can act as a potential tool for the diagnosis of HCV
infection.
To achieve the greatest possible sensitivity and
specificity, we chose two formats of gold-based immunoassays. In
the double antigen sandwich immunoassay, the HCV antigen labelled
with colloidal gold is a soluble recombinant antigen. During the
test, HCV antibodies in the sample react with antigen coated on the
nitrocellucose membrane and gold-HCV antigen conjugates, forming a
gold-conjugated Ag-Ab-Ag sandwich complex. At the same time, we
carried out a series of experiments to optimize different
parameters, such as the amount of immunoreagents, the type of
materials, and the composition of the blocking solution and
detector reagent mixture. The experimental results demonstrated
that the chromatographic strip device we constructed is simple,
sensitive and specific. It is an ideal test for the screening of
patients with HCV infection. The presence of recombinant core and
NS3 antigens in the strip may be crucial for the detection of
HCV-infected patients with low antibody titers.
In conclusion, we found that recombinant antigens
encoded by different HCV gene fragments display different
immunoreactivity to anti-HCV antibodies. Our data show the
full-length core and NS3 (1183–1476 aa) proteins have the major
immuno-dominant epitopes of the HCV genome. More importantly, our
study verify that the full-length core and NS3 (1183-1476 aa)
recombinant antigen can be used to construct a double antigen
sandwich lateral-flow immunochromatographic anti-HCV immunoassay
strip. This strip allows for the more rapid and more economical
detection of HCV. It also has a high sensitivity and specificity in
testing for HCV. It has the potential to become a useful tool for
HCV clinical detection.
Acknowledgements
This study was financially supported
by Artron BioResearch Inc. and the Chongqing Natural Science
Foundation (#CSTC, 2010AB5012). The authors would like to thank Dr
XueFei Cai (Chongqing Medical University) for providing pET-C33 and
pET-C22.
References
|
1.
|
J NakamuraK TerajimaY AoyagiK
AkazawaCost-effectiveness of the national screening program for
hepatitis C virus in the general population and the high-risk
groupsTohoku J Exp Med2153342200810.1620/tjem.215.3318509233
|
|
2.
|
L BelloniF MorettiP MerloDNp73alpha
protects myogenic cells from
apoptosisOncogene2536063612200610.1038/sj.onc.120932116652159
|
|
3.
|
D SchuppanA KrebsM BauerEG HahnHepatitis C
and liver fibrosisCell Death Differ10Suppl
1S59S67200310.1038/sj.cdd.4401163
|
|
4.
|
JC LuoSJ HwangCP LiClinical significance
of serum auto-antibodies in Chinese patients with chronic hepatitis
C: negative role of serum viral titre and genotypeJ Gastroenterol
Hepatol13475479199810.1111/j.1440-1746.1998.tb00671.x9641643
|
|
5.
|
M SchroeterB ZoellnerS PolywkaR LaufsHH
FeuchtProlonged time until seroconversion among hemodialysis
patients: the need for HCV
PCRIntervirology48213215200510.1159/00008459715920344
|
|
6.
|
HL ZaaijerHT CuypersHW ReesinkIN WinkelG
GerkenPN LelieReliability of polymerase chain reaction for
detection of hepatitis C
virusLancet341722724199310.1016/0140-6736(93)90488-38095626
|
|
7.
|
K ShahzamaniF SabahiS MeratRapid low-cost
detection of hepatitis C virus RNA in HCV-infected patients by
real-time RT-PCR using SYBR Green IArch Iran
Med14396400201122039844
|
|
8.
|
G MachnikE PelcM ZapalaDesigning and
optimization of real-time RT-PCR technique for the detection of
hepatitis C virus (HCV) genome in blood serum as internal
laboratory quality controlPrzegl Epidemiol653253322011(In
Polish).
|
|
9.
|
Q GaoD LiuS ZhangL TongAnalyses of
anti-hCV detected by ELISA and HCV RNA detected by RT-nPCR in
chronic hepatitis C virus infectorsWei Sheng Yan Jiu3669712007(In
Chinese).
|
|
10.
|
AK ReddyKV DakshinamurtyV LakshmiUtility
of HCV core antigen ELISA in the screening for hepatitis C virus
infection in patients on hemodialysisIndian J Med
Microbiol245557200610.4103/0255-0857.1989716505558
|
|
11.
|
M RiosM DiagoP RiveraEpidemiological,
biological and histological characterization of patients with
indeterminate third-generation recombinant immunoblot assay
antibody results for hepatitis C virusJ Viral
Hepat13177181200610.1111/j.1365-2893.2005.00673.x
|
|
12.
|
HD TungSN LuCM LeeAntiviral treatment
responses in patients with chronic hepatitis C virus infection
evaluated by a third generation anti-hepatitis C virus assayJ Viral
Hepat9304308200210.1046/j.1365-2893.2002.00359.x12081608
|
|
13.
|
S SookoianG CastanoEvaluation of a third
generation anti-HCV assay in predicting viremia in patients with
positive HCV antibodiesAnn Hepatol1179182200215280804
|
|
14.
|
J ZhangD WangY LiSARS coronavirus
nucleocapsid protein monoclonal antibodies developed using a
prokaryotic expressed proteinHybridoma
(Larchmt)30481485201110.1089/hyb.2011.0028
|
|
15.
|
T GoeserHM MullerJ YeE PfaffL
TheilmannCharacterization of antigenic determinants in the core
antigen of the hepatitis C
virusVirology205462469199410.1006/viro.1994.16667526540
|
|
16.
|
M BeldM PenningM van PuttenQuantitative
antibody responses to structural (Core) and nonstructural (NS3,
NS4, and NS5) hepatitis C virus proteins among seroconverting
injecting drug users: impact of epitope variation and relationship
to detection of HCV RNA in
bloodHepatology2912881298199910.1002/hep.510290442
|
|
17.
|
SJ HwangHepatitis C virus infection: an
overviewJ Microbiol Immunol Infect34227234200111825001
|
|
18.
|
LI NikolaevaNP BlokhinaNN
TsurikovaVirus-specific antibody titres in different phases of
hepatitis C virus infectionJ Viral
Hepat9429437200210.1046/j.1365-2893.2002.00369.x12431205
|
|
19.
|
C Jolivet-ReynaudA AdidaS
MichelCharacterization of mimotopes mimicking an immunodominant
conformational epitope on the hepatitis C virus NS3 helicaseJ Med
Virol72385395200410.1002/jmv.2000214748062
|
|
20.
|
T KatoM MiyamotoA FurusakaProcessing of
hepatitis C virus core protein is regulated by its C-terminal
sequenceJ Med Virol69357366200310.1002/jmv.1029712526046
|
|
21.
|
HH FeuchtB ZollnerS PolywkaR LaufsStudy on
reliability of commercially available hepatitis C virus antibody
testsJ Clin Microbiol3362062419957751366
|