Introduction
Cell adhesion molecules are cellular surface
proteins involved in binding with other cells or with the
extracellular matrix (ECM) in a process called cell adhesion. These
molecules play a critical role in the inflammatory response and
tumor progression, which promotes cancer-defining biological
processes such growth, survival, migration, and metastasis.
Therefore, cell adhesion molecule expression is essential for
maintaining cell homeostasis. Among the cell adhesion molecules,
intercellular adhesion molecule (ICAM-1) and vascular cell adhesion
molecule (VCAM-1) are constitutively expressed on airway
endothelial and epithelial barriers during inflammation, which may
play a key role in the recruitment and infiltration of leukocytes
across the blood vessels at sites of airway inflammation (1–3).
ICAM-1 and VCAM-1 have been shown to be upregulated in vascular
cells by several pro-inflammatory cytokines such as tumor necrosis
factor-α (TNF-α) (4–6).
Nuclear factor-κB (NF-κB) is a well-known
transcription factor that regulates the expression of cell adhesion
molecules and the development of inflammatory responses by
upregulating inflammatory mediators (7,8).
Previous reports suggest that TNF-α activates the NF-κB signaling
pathway, which results in the upregulation of cell adhesion
molecules, including ICMA-1 and VCAM-1 (9,10).
Inhibition of NF-κB activation induces the suppression of
TNF-α-induced ICAM-1 and VCAM-1 mRNA and protein expression levels
(11). Therefore, NF-κB, the
central regulator in adhesion molecule expression, is a more
effective target for anti-inflammatory therapy.
Generally regarded as safe, natural compounds have
been shown to have antitumor, anti-angiogenic and anti-inflammatory
effects against several cell types (12). Butein
(3,3,2′,4′-tetrahydroxychalcone) is a biologically active
polyphenol compound derived from numerous plants including the
stembark of cashews (Semecarpus anacardium), the heartwood
of Dalbergia odorifera, and the traditional Chinese
medicinal herbs Caragana jubata and Rhus verniciflua
Stokes. Several studies have reported its anti-carcinogenic
activities by inhibiting the proliferation of a wide variety of
tumor cells (13). Butein can
induce growth inhibition through G2/M phase arrest in hepatic cells
(14). Butein induces apoptosis
through the suppression of STAT-3 gene expression (13) and the sensitization of human
hepatoma cells to TRAIL-induced apoptosis via DR5 upregulation and
promote NF-κB inactivation (15).
Recently, it has been reported that butein inhibits the expression
of CXC chemokine receptor-4 (CXCR4), a mediator of the growth and
metastasis of tumors, and thus has the potential to suppress cancer
metastasis (16).
Despite evidence that butein can block tumor cell
growth and have anti-proliferative activities, the effect of butein
on the expression level of specific adhesion molecules (ICAM-1 and
VCAM-1) and its anti-inflammatory molecular mechanism have not been
elucidated. Therefore, we investigated the effects and the
molecular mechanism of butein on TNF-α-induced adhesion molecule
expression in human lung epithelial cells. We found that butein
inhibited TNF-α-induced ICAM-1 and VCAM-1 expression and monocyte
adhesion through the inhibition of NF-κB, MAPK and Akt signaling
pathways and reactive oxygen species (ROS) generation.
Materials and methods
Cell culture and materials
Human lung epithelial A549 cells and human monocyte
leukemia U937 cells were obtained from the ATCC (Rockville, MD,
USA). The cells were cultured in RPMI-1640 supplemented with 2 mM
L-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin,
and 10% FBS. The cells were grown at 37°C and 5% CO2 in
95% humidified air and subcultured twice weekly. Human recombinant
TNF-α was obtained from R&D Systems (Minneapolis, MN, USA).
Anti-VACM-1 and anti-ICAM-1 antibodies were purchased from Santa
Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). Anti-phospho-ERK,
anti-phospho-JNK, anti-phospho-p38 MAPK, anti-phospho-Akt and
anti-NF-κB-p65 antibodies were purchased from Cell Signaling
Technology, Inc. (Boston, MA, USA). PD98059 (ERK inhibitor),
SP600125 (JNK inhibitor), and SB203580 (p38 inhibitor) were
purchased from Calbiochem (San Diego, CA, USA). Butein and all
other reagents were purchased from Sigma-Aldrich.
Western blot analysis
Cells were collected and washed twice with ice-cold
phosphate-buffered saline (PBS) pelleted at 1,000 × g for 5 min.
Cellular lysates were prepared by suspending 1×106 cells
in 100 μl of ice-cold lysis buffer (137 mM NaCl, 15 mM EGTA,
0.1 mM sodium orthovanadate, 15 mM MgCl2, 0.1% Triton
X-100, 25 mM MOPS, 100 μM phenylmethylsulfonyl fluoride, and
20 μM leupeptin, adjusted to pH 7.2) for 20 min on ice. The
extracts were centrifuged at 12,000 × g for 20 min at 4°C. The
protein content in the supernatant was measured using a Bio-Rad
Protein Assay kit (Bio-Rad, Hercules, CA, USA). The protein lysates
were then denatured at 95°C for 5 min after mixing with 5X
SDS-loading buffer and loaded on an SDS polyacrylamide gel for
electrophoresis. The proteins were then transferred to Immobilon-P
membranes (Millipore Corporation, Bedford, MA, USA). The detection
of specific proteins was carried out with an ECL western blotting
kit (Millipore Corporation and Amersham Biosciences,
Buchinghamshire, UK) according to the manufacturer’s
instructions.
Cell adhesion assay
A549 cells were grown to 80–90% confluence in 35-mm
plates. Monocyte U937 cells were incubated in RPMI-1640 medium
containing 2% FBS and 10 μg/ml of the fluorescent dye
BCECF/AM (485 nm excitation/535 nm emission wavelengths;
Calbiochem) at 37°C for 30 min. After stimulating the A549 cells
with 10 ng/ml of TNF-α for 6 h following pretreatment with or
without 5 µM of butein for 30 min, the cells were washed
three times with PBS and re-suspended in phenol red-free RPMI-1640
containing 2% FBS, and fluorescently labeled U937 cells were added
(1×106 cells/ml). After incubation for 30 min at 37°C,
non-adherent U937 cells were removed by washing three times with
PBS. U937 cell adhesion was observed under a fluorescence
microscope and then counted in 5 randomly selected microscopic
fields in each well.
RNA isolation and reverse
transcriptase-polymerase chain reaction (RT-PCR)
Total RNA was isolated as previously described
(17). Single-strand cDNA was
synthesized from 2 μg of total RNA using Moloney murine
leukemia virus (MMLV) reverse transcriptase (Gibco-BRL,
Gaithersburg, MD, USA). The cDNAs for ICAM-1, VCAM-1 and actin were
amplified by PCR with specific primers. The sequences of the
forward and reverse primers were, respectively: ICAM-1, 5′-AGA GAT
GAC CAT GGA GCC-3′ and 5′-TCC CTT CTG AGA CCT CTG-3′; VCAM-1,
5′-GAA CAC TCT TAC CTG TGC ACA GCA AC-3′ and 5′-GGA GCT GGT AGA CCC
TCG CTG G-3′; actin, 5′-GGC ATC GTC ACC AAC TGG GAC-3′ and 5′-CGA
TTT CCC GCT CGG CCG TGG-3′. The conditions for the PCR reaction
were as follows: 26 cycles of 95°C for 45 sec, 56°C for 45 sec, and
72°C for 45 sec for ICAM-1; 26 cycles of 95°C for 45 sec, 62°C for
45 sec, and 72°C for 45 sec for VCAM-1; and 17 cycles of 95°C for
30 sec, 56°C for 30 sec, and 72°C for 35 sec for actin. PCR
products were analyzed by agarose gel electrophoresis and
visualized by ethidium bromide.
Luciferase reporter gene assay
The NF-κB reporter construct was purchased from
Clontech (Palo Alto, CA, USA). ICAM-1 and VCAM-1 luciferase
plasmids containing regions spanning from −1350 to +45 bp (full
length) of the human ICAM-1 promoter and regions spanning −1716 to
+119 bp (full length) of the human VCAM-1 promoter were used as
previously reported (18). NF-κB
reporter plasmids or ICAM-1 or VCAM-1 promoter plasmids were
transfected into A549 cells using the Lipofectamine reagent
according to the manufacturer’s instructions. To assess NF-κB,
ICAM-1 or VCAM-1 promoter activity, cells were collected and
disrupted by sonication in lysis buffer (25 mM Tris-phosphate, pH
7.8, 2 mM EDTA, 1% Triton X-100, and 10% glycerol). Following
centrifugation, aliquots of the supernatants were analyzed with a
luciferase assay according to the manufacturer’s instructions.
Assessment of NF-κB-p65-EGFP nuclear
translocation
A549 cells were seeded and transfected with
NF-κB-p65-EGFP vector (provided by Dr Jun C.D., GIST, Korea). After
24 h of transfection, A549 cells were pretreated with 0.5 μM
of butein for 1 h at 37°C and then exposed to 10 ng/ml of TNF-α for
1 h. The cells were fixed with 1% paraformaldehyde on glass slides
for 30 min at room temperature. After washing with PBS, 300 nM of
DAPI (4′-6′-diamidino-2-phenylindole; Roche, Germany) was added to
the fixed cells for 5 min. The slides were washed and air dried
before they were mounted with coverslips with ProLong®
antifade mounting medium (Molecular Probes, USA), and they were
subsequently examined using a fluorescence microscope. Fluorescence
images were observed using a confocal laser scanning microscope
(LSM 5 Exciter; Carl Zeiss, Jena, Germany) connected to an
Axio-observer Z1 inverted microscope using a C-Apochromat ×40
objective. Image analysis was completed with LSM 5 Exciter software
(Carl Zeiss).
Measurement of reactive oxygen
species
A549 cells were plated in 6-well plates at a density
of 4×105 cells/ml, allowed to attach overnight, and
treated with TNF-α (10 ng/ml), butein (5 µM), or TNF-α and
butein in combination. Intracellular hydrogen peroxide levels were
detected by flow cytometry using 2′,7′-dichlorodihydrofluorescein
diacetate (H2DCFDA) (19). This dye is a stable, nonpolar
compound that readily diffuses into cells and is hydrolyzed by
intracellular esterase to yield DCFH. Hydrogen peroxide produced by
cells oxidizes DCFH to the highly fluorescent compound
2′,7′-dichlorofluorescein (DCF). After drug treatment, the cells
were treated with 5 μM of H2DCFDA and then
incubated at 37°C for 20 min. The cells were harvested and washed
twice with PBS, and the fraction of DCF-positive cells was observed
under a fluorescence microscope (Axiovert 200M; Carl Zeiss).
Statistical analysis
Results are represented as the means ± SD. The
difference between two mean values was analyzed by the Student’s
t-test and was considered to be statistically significant at
P<0.05.
Results
Butein inhibits TNF-α-induced monocyte
adhesion to A549 cells
Adhesion of monocytes to endothelial cells is an
important step for the initiation and promotion of the inflammatory
process. To investigate the effect of butein on the adhesion of
monocytes to TNF-α-treated lung epithelial cells, we performed a
monocyte adhesion assay. A549 cells were pretreated with or without
butein for 30 min before incubation with TNF-α (10 ng/ml) for 6 h,
and then co-cultured with fluorescently labeled human U937 cells.
TNF-α treatment significantly increased the adhesion of U937 cells
to A549 cells by 6-fold compared with the untreated control cells
(Fig. 1A). However, butein
significantly inhibited TNF-α-induced monocyte adhesion to A549
cells in a dose-dependent manner, and maximum inhibition was
estimated to be ∼70% with 5 µM butein pretreatment (Fig. 1A). Butein and TNF-α did not affect
cellular cytotoxicity when compared with the control cells over a
24 h incubation period. Cell viability remained >98% in all
groups assessed by the MTT assay (Fig. 1B).
Butein suppresses the TNF-α-induced
expression of ICAM-1 and VCAM-1 in A549 cells
The expression of cell adhesion molecules is related
to monocyte adhesion to epithelial cells during inflammation
(2). ICAM-1 and VCAM-1 mRNA and
protein expression levels were assessed to investigate whether the
inhibitory effect of butein on TNF-α-induced monocyte adhesion was
due to the downregulation of adhesion molecules. A549 cells were
incubated with or without various concentrations of butein for 30
min prior to induction with TNF-α (10 ng/ml) for 12 h. TNF-α
significantly increased the protein expression of ICAM-1 and VCAM-1
in A549 cells compared to control-treated cells, and butein
effectively inhibited the expression of these molecules in a
dose-dependent manner (Fig. 2A).
Next, we examined the effect of butein on TNF-α-induced ICAM-1 and
VCAM-1 transcription using RT-PCR analysis. ICAM-1 and VCAM-1 mRNA
were expressed at low levels in unstimulated and butein-treated
A549 cells, but there was a marked increase in mRNA expression in
cells stimulated with TNF-α. Butein significantly suppressed the
TNF-α-induced increase in ICAM-1 and VCAM-1 mRNA levels in a
dose-dependent manner (Fig. 2B).
The effect of butein on transcriptional regulation was further
investigated by assaying ICAM-1 and VCAM-1 promoter activities.
Butein markedly reduced the TNF-α-induced luciferase activity in a
dose-dependent manner (Fig. 2C and
D). These results suggest that butein suppresses ICAM-1 and
VCAM-1 mRNA expression and thereby reduces monocyte adhesion to
epithelial cells.
Butein inhibits TNF-α-induced NF-κB
activation
Previous studies have shown that the signaling
pathway of NF-κB, a well known transcription factor, is involved in
the gene expression of various proteins involved in the
inflammatory response, including the cell adhesion molecules ICAM-1
and VCAM-1 (20). To confirm the
effect of butein on TNF-α-induced NF-κB-dependent reporter gene
expression, we used an NF-κB-luciferase construct, which was
generated by introducing four NF-κB binding sites into the
pLuc-promoter vector. We transiently transfected A549 cells with
the pNF-κB-Luc plasmid and stimulated the cells with TNF-α (10
ng/ml) either in the presence or absence of butein. An ∼4-fold
increase in NF-κB luciferase activity was detected after
stimulation with TNF-α for 12 h. Butein significantly reduced the
TNF-α-induced NF-κB luciferase activity (Fig. 3A). In the unstimulated state,
NF-κB is located in the cytosol and is complexed with the
inhibitory protein IκBα. The dissociation of IκBα from NF-κB
results in NF-κB activation (21). Therefore, we also performed
western blotting to detect the translocation of the NF-κB-p65
subunit. Butein inhibited NF-κB-p65 translocation to the nuclear
fraction in a dose-dependent manner (Fig. 3B). In addition, we further
analyzed the nuclear translocation of the NF-κB-p65 subunit by
immunofluorescence. A549 cells were transfected with the
NF-κB-p65-EGFP vector for 24 h and co-treated with butein and
TNF-α. We found that butein prevented the TNF-α-induced nuclear
translocation of the NF-κB-p65 subunit (Fig. 3C). Therefore, these results
suggest that butein inhibits NF-κB activation through the
suppression of TNF-α-induced nuclear translocation of NF-κB in A549
cells.
Butein regulates the TNF-α-induced
activation of the MAPK and Akt signaling pathways
Since the MAPK pathways are involved in controlling
a wide range of cellular responses including growth,
differentiation, inflammation and apoptosis, we examined the
activation of MAPKs by western blotting using specific
anti-phospho-kinase antibodies. TNF-α clearly increased the levels
of JNK, p38 MAPK and ERK phosphorylation compared with untreated
cells. The TNF-α-induced MAPK activation was significantly
inhibited by pretreatment with butein (Fig. 4A). To confirm whether MAPK
signaling pathways are involved in TNF-α-induced ICAM-1 and VCAM-1
expression, we used specific inhibitors of JNK (SP600125), p38 MAPK
(SB203580) and ERK (PD98059) on TNF-α-stimulated A549 cells.
Similar to our earlier results (Fig.
2), pretreatment with inhibitors significantly decreased
TNF-α-induced ICAM-1 and VCAM-1 expression (Fig. 4B). ICAM-1 was inhibited by
treatment with either PD98059 or SP600125. TNF-α-induced VCAM-1
expression was upregulated by either ERK or JNK inhibition.
Notably, VCAM-1 was specifically inhibited by treatment with
SB203580.
Akt [protein kinase B (PKB)] is a serine/threonine
protein kinase that plays a key role in adhesion molecule
expression in TNF-α-induced epithelial cells (22). To investigate whether butein
affects TNF-α-induced Akt activation, we examined the effect of
butein on the TNF-α-induced Akt phosphorylation in A549 cells using
western blot analysis. While Akt activation was gradually increased
in TNF-α-treated A549 cells, a 10-min butein treatment completely
blocked the increase of TNF-α-induced Akt phosphorylation (Fig. 4C). These results suggest that
butein may reduce TNF-α-induced ICAM-1 and VCAM-1 expression by
suppressing the phosphorylation of the MAPK and Akt pathway.
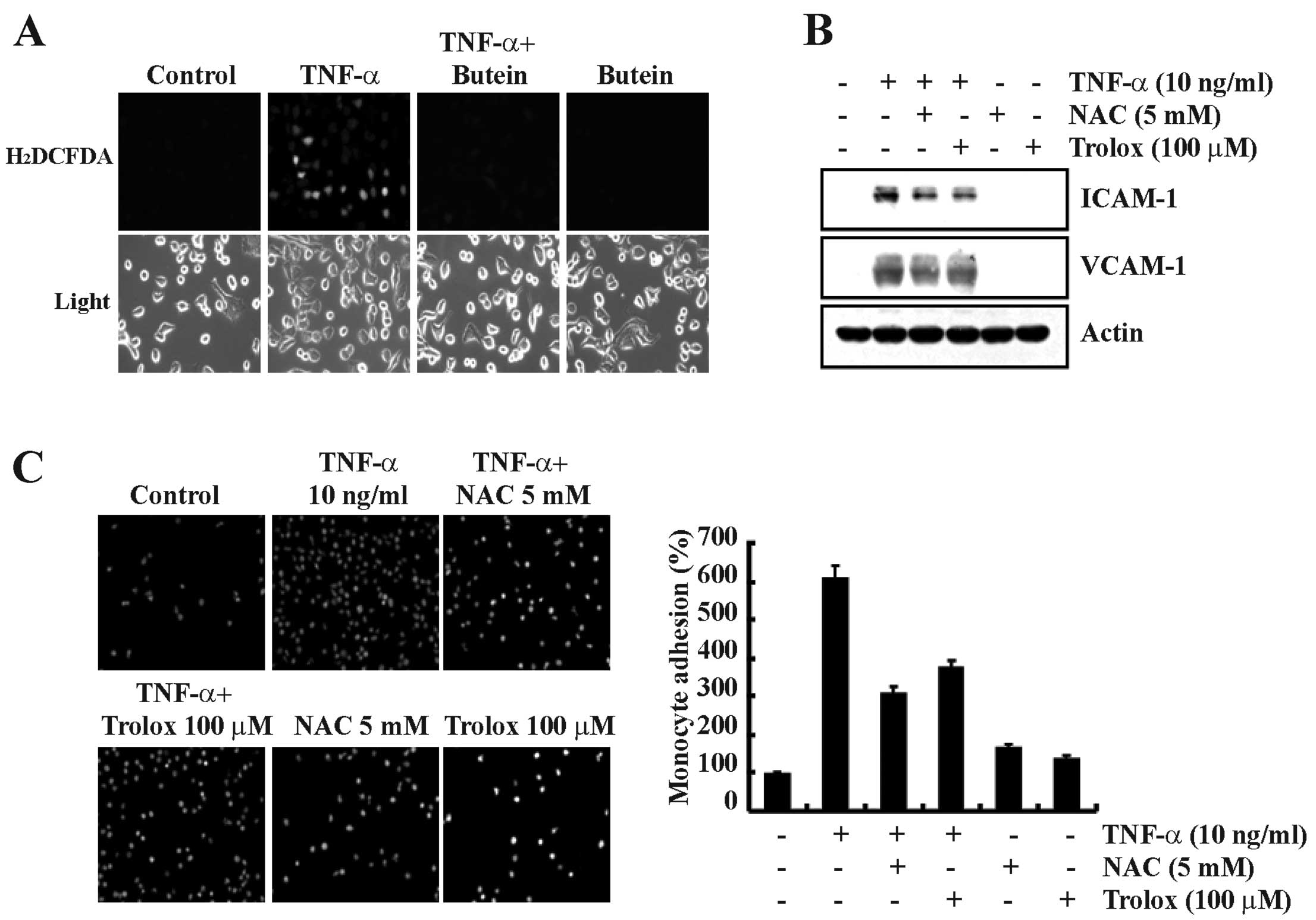
Butein inhibits TNF-α-induced ROS
generation in A549 cells
ROS are important signaling molecules associated
with the expression of cell adhesion molecules in response to TNF-α
(23). We also found that butein
inhibits TNF-α-induced NF-κB activation (Fig. 3). Therefore, we examined the
effect of butein on TNF-α-induced ROS generation in A549 cells.
Butein significantly inhibited the production of TNF-α-induced
hydrogen peroxide levels, as visualized by fluorescence microscopy
(Fig. 5A). To confirm whether ROS
generation was involved in TNF-α-induced ICAM-1 and VCAM-1
expression, we examined the effect of the antioxidants NAC
(N-acetylcysteine) and Trolox on TNF-α-stimulated A549 cells. These
antioxidant molecules effectively inhibited TNF-α-induced ICAM-1
and VCAM-1 expression (Fig. 5B).
In addition, TNF-α-induced monocyte adhesion to A549 cells was also
inhibited by treatment with the antioxidant molecules (Fig. 5C). These results suggest that the
inhibition of TNF-α-induced ICAM-1 and VCAM-1 expression by butein
may be mediated by the inhibition of ROS generation.
Discussion
The inhibitory effect of many flavonoids on the
expression of adhesion molecules is expected to have an
anti-inflammatory effect by decreasing leukocyte recruitment. In
the present study, we investigated whether butein, an anticancer
drug derived from natural sources, is associated with
anti-inflammatory activities. Our results showed that butein
significantly inhibited both ICAM-1 and VCAM-1 expression levels in
TNF-α-stimulated A549 cells, and these effects potentially
modulated monocyte adhesion to A549 cells. We found that the
inhibitory effect of butein on TNF-α-treated A549 cells was
mediated through the inhibition of NF-κB activation, Akt
phosphorylation and ROS generation.
Cell adhesion molecule expression has been reported
to be associated with airway inflammation and the migration and
recruitment of lymphocytes (24).
Numerous studies have shown that TNF-α, a pro-inflammatory
cytokine, can induce the expression of adhesion molecules,
including ICAM-1 and VCAM-1, which contribute to the inflammatory
process (25). We also found that
TNF-α induced the expression of adhesion molecules (ICAM-1 and
VCAM-1) and increased monocyte adhesion to A549 cells, which were
significantly inhibited by treatment with butein in a
dose-dependent manner (Figs. 1
and 2). Our results suggest that
butein can prevent TNF-α-induced airway inflammation.
NF-κB is known to play an important role in the
rapid induction of a variety of cytokines (TNF-α, MCP-1) and the
expression of adhesion molecules (ICAM-1 and VCAM-1) (26). Previous studies have also shown
that NF-κB inhibitory agents (PDTC, curcumin and salicylates)
suppress the expression of cell adhesion molecules (11,27,28). In this study, we found that butein
significantly inhibited TNF-α-induced NF-κB promoter activity and
the nuclear translocation of NF-κB-p65 (Fig. 3). Therefore, butein may be a
potential therapeutic agent for suppressing the NF-κB-dependent
inflammatory response.
Several reports suggest that the MAPK (JNK, p38 and
ERK) signaling pathways are differentially involved in the
anti-inflammatory response and the TNF-α-induced expression of
adhesion molecules (29,30). Akt has been shown to be involved
in NF-κB activation and adhesion molecule expression induced by
TNF-α (31). Moreover, butein can
attenuate ERK and p38 MAPK activation to block NF-κB signaling in
human cancer cells (32,33). Therefore, we investigated the
effect of butein on TNF-α- induced MAPK and Akt signaling pathway.
Our results showed that butein significantly inhibited
TNF-α-induced Akt phosphorylation and MAPK phosphorylation in lung
epithelial A549 cells (Fig. 4).
In addition, we previously reported that PI3K/Akt specific
inhibitor, LY294002, inhibits TNF-α-induced cell adhesion and
VCAM-1 expression (34).
Therefore, these results suggest that butein may inhibit
TNF-α-induced ICAM-1 and VCAM-1 expression through the inhibition
of Akt and MAPK activation.
Reactive oxygen species (ROS), such as the
superoxide anion radical (O2−), the hydroxyl
radical (·OH) and hydrogen peroxide (H2O2),
are generated as a result of incomplete reaction of oxygen during
aerobic metabolism or environmental stresses such as radiation, UV,
heat and several oxidants (35).
Many inflammatory cytokines such as TNF-α may generate ROS
(36). Adhesion molecule
expression has been implicated to occur as a result of
ROS-dependent NF-κB activation, and ROS scavenging agents inhibit
the ROS-mediated activation of NF-κB (37,38). Furthermore, multiple biological
properties of polyphenol compounds have been attributed to their
antioxidant functions through their influence on intracellular
redox status (39). Therefore, we
studied the effect of butein on TNF-α-induced ROS generation. Our
study showed that butein effectively inhibits TNF-α-induced ROS
generation in A549 cells, which results in the inhibition of ICAM-1
and VCAM-1 expression (Fig. 5).
The inhibitory effect of butein on TNF-α-induced adhesion molecule
expression is due to the inhibition of intracellular ROS generation
and relevant signaling pathways.
In conclusion, the present study demonstrated that
butein is capable of inhibiting the expression of ICAM-1 and VCAM-1
in TNF-α-stimulated A549 cells. The inhibition of adhesion molecule
(ICAM-1 and VCAM-1) expression subsequently inhibited monocyte
adhesion to TNF-α-stimulated A549 cells. We found that the
inhibitory effect of butein is mediated through the inhibition of
NF-κB, Akt and MAPK activation, and the inhibition of ROS
generation also contributed to the inhibition of ICAM-1 and VCAM-1
expression. Thus, butein could be useful in the development of
therapeutic drugs against various inflammatory diseases.
References
|
1.
|
AJ PolitoD ProudEpithelia cells as
regulators of airway inflammationJ Allergy Clin
Immunol102714718199810.1016/S0091-6749(98)70008-99867498
|
|
2.
|
S RosseauJ SelhorstK WiechmannMonocyte
migration through the alveolar epithelial barrier: adhesion
molecule mechanisms and impact of chemokinesJ
Immunol164427435200010.4049/jimmunol.164.1.42710605039
|
|
3.
|
E RijckenCF KrieglsteinC AnthoniICAM-1 and
VCAM-1 antisense oligonucleotides attenuate in vivo leucocyte
adherence and inflammation in rat inflammatory bowel
diseaseGut51529535200210.1136/gut.51.4.52912235075
|
|
4.
|
A Burke-GaffneyPG HellewellTumour necrosis
factor-alpha-induced ICAM-1 expression in human vascular
endothelial and lung epithelial cells: modulation by tyrosine
kinase inhibitorsBr J
Pharmacol11911491158199610.1111/j.1476-5381.1996.tb16017.x
|
|
5.
|
MI CybulskyK IiyamaH LiA major role for
VCAM-1, but not ICAM-1, in early atherosclerosisJ Clin
Invest10712551262200110.1172/JCI1187111375415
|
|
6.
|
Y HuoK LeyAdhesion molecules and
atherogenesisActa Physiol
Scand1733543200110.1046/j.1365-201X.2001.00882.x11678724
|
|
7.
|
S GhoshMJ MayEB KoppNF-kappa B and Rel
proteins: evolutionarily conserved mediators of immune
responsesAnnu Rev
Immunol16225260199810.1146/annurev.immunol.16.1.2259597130
|
|
8.
|
AS Baldwin JrSeries introduction: the
transcription factor NF-kappaB and human diseaseJ Clin
Invest10736200110.1172/JCI1189111134170
|
|
9.
|
AA BegTS FincoPV NantermetAS Baldwin
JrTumor necrosis factor and interleukin-1 lead to phosphorylation
and loss of I kappa B alpha: a mechanism for NF-kappa B
activationMol Cell Biol133301331019938497253
|
|
10.
|
CC ChenCL RosenbloomDC AndersonAM
ManningSelective inhibition of E-selectin, vascular cell adhesion
molecule-1, and intercellular adhesion molecule-1 expression by
inhibitors of I kappa B-alpha phosphorylationJ
Immunol155353835451995
|
|
11.
|
JW PierceMA ReadH DingFW LuscinskasT
CollinsSalicylates inhibit I kappa B-alpha phosphorylation,
endothelialleukocyte adhesion molecule expression, and neutrophil
transmigrationJ Immunol156396139691996
|
|
12.
|
DJ NewmanNatural products as leads to
potential drugs: an old process or the new hope for drug
discovery?J Med Chem5125892599200810.1021/jm070409018393402
|
|
13.
|
MK PandeyB SungKS AhnBB AggarwalButein
suppresses constitutive and inducible signal transducer and
activator of transcription (STAT) 3 activation and STAT3-regulated
gene products through the induction of a protein tyrosine
phosphatase SHP-1Mol
Pharmacol75525533200910.1124/mol.108.05254819103760
|
|
14.
|
DO MoonMO KimYH ChoiJW HyunWY ChangGY
KimButein induces G(2)/M phase arrest and apoptosis in human
hepatoma cancer cells through ROS generationCancer
Lett288204213201010.1016/j.canlet.2009.07.00219643530
|
|
15.
|
DO MoonMO KimYH ChoiGY KimButein
sensitizes human hepatoma cells to TRAIL-induced apoptosis via
extracellular signal-regulated kinase/Sp1-dependent DR5
upregulation and NF-kappaB inactivationMol Cancer
Ther915831595201010.1158/1535-7163.MCT-09-0942
|
|
16.
|
AW ChuaHS HayP RajendranButein
downregulates chemokine receptor CXCR4 expression and function
through suppression of NF-kappaB activation in breast and
pancreatic tumor cellsBiochem
Pharmacol8015531562201010.1016/j.bcp.2010.07.04520699088
|
|
17.
|
P ChomczynskiN SacchiSingle-step method of
RNA isolation by acid guanidinium thiocyanate-phenol-chloroform
extractionAnal
Biochem162156159198710.1016/0003-2697(87)90021-22440339
|
|
18.
|
I KimSO MoonSH KimHJ KimYS KohGY
KohVascular endothelial growth factor expression of intercellular
adhesion molecule 1 (ICAM-1), vascular cell adhesion molecule 1
(VCAM-1), and E-selectin through nuclear factor-kappa B activation
in endothelial cellsJ Biol
Chem27676147620200110.1074/jbc.M009705200
|
|
19.
|
B ZeguraTT LahM FilipicThe role of
reactive oxygen species in microcystin-LR-induced DNA
damageToxicology2005968200410.1016/j.tox.2004.03.00615158564
|
|
20.
|
CK GlassJL WitztumAtherosclerosis: the
road
aheadCell104503516200110.1016/S0092-8674(01)00238-011239408
|
|
21.
|
MS RodriguezJ ThompsonRT HayC
DargemontNuclear retention of IkappaBalpha protects it from
signal-induced degradation and inhibits nuclear factor kappaB
transcriptional activationJ Biol
Chem27491089115199910.1074/jbc.274.13.9108
|
|
22.
|
JK MinYM KimSW KimTNF-related
activation-induced cytokine enhances leukocyte adhesiveness:
induction of ICAM-1 and VCAM-1 via TNF receptor-associated factor
and protein kinase C-dependent NF-kappaB activation in endothelial
cellsJ Immunol175531540200510.4049/jimmunol.175.1.531
|
|
23.
|
H KimJS HwangCH WooTNF-alpha-induced
upregulation of intercellular adhesion molecule-1 is regulated by a
Rac-ROS-dependent cascade in human airway epithelial cellsExp Mol
Med40167175200810.3858/emm.2008.40.2.16718446055
|
|
24.
|
SM AlbeldaCW SmithPA WardAdhesion
molecules and inflammatory injuryFASEB J850451219948181668
|
|
25.
|
G LeeHJ NaS Namkoong4-O-methylgallic acid
downregulates endothelial adhesion molecule expression by
inhibiting NF-kappaB-DNA-binding activityEur J
Pharmacol551143151200610.1016/j.ejphar.2006.08.06117027748
|
|
26.
|
PP TakGS FiresteinNF-kappaB: a key role in
inflammatory diseasesJ Clin
Invest107711200110.1172/JCI1183011134171
|
|
27.
|
N MaruiMK OffermannR SwerlickVascular cell
adhesion molecule-1 (VCAM-1) gene transcription and expression are
regulated through an antioxidant-sensitive mechanism in human
vascular endothelial cellsJ Clin
Invest9218661874199310.1172/JCI116778
|
|
28.
|
S SinghBB AggarwalActivation of
transcription factor NF-kappa B is suppressed by curcumin
(diferuloylmethane) [corrected]J Biol Chem27024995250001995
|
|
29.
|
AW HoCK WongCW LamTumor necrosis
factor-alpha upregulates the expression of CCL2 and adhesion
molecules of human proximal tubular epithelial cells through MAPK
signaling
pathwaysImmunobiology213533544200810.1016/j.imbio.2008.01.003
|
|
30.
|
JW JuSJ KimCD JunJS Chunp38 kinase and
c-Jun N-terminal kinase oppositely regulates tumor necrosis factor
alpha-induced vascular cell adhesion molecule-1 expression and cell
adhesion in chondrosarcoma cellsIUBMB
Life54293299200210.1080/15216540215674
|
|
31.
|
JS KangYD YoonMH HanGlabridin suppresses
intercellular adhesion molecule-1 expression in tumor necrosis
factor-alpha-stimulated human umbilical vein endothelial cells by
blocking sphingosine kinase pathway: implications of Akt,
extracellular signal-regulated kinase, and nuclear
factor-kappaB/Rel signaling pathwaysMol Pharmacol699419492006
|
|
32.
|
L ZhangW ChenX LiA novel anticancer effect
of butein: inhibition of invasion through the ERK1/2 and NF-kappa B
signaling pathways in bladder cancer cellsFEBS
Lett58218211828200810.1016/j.febslet.2008.04.04618472007
|
|
33.
|
SH LeeGS SeoXY JinG KoDH SohnButein blocks
tumor necrosis factor alpha-induced interleukin 8 and matrix
metalloproteinase 7 production by inhibiting p38 kinase and
osteopontin mediated signaling events in HT-29 cellsLife
Sci8115351543200710.1016/j.lfs.2007.09.024
|
|
34.
|
JH OhTK KwonWithaferin A inhibits tumor
necrosis factor-alpha-induced expression of cell adhesion molecules
by inactivation of Akt and NF-kappaB in human pulmonary epithelial
cellsInt
Immunopharmacol9614619200910.1016/j.intimp.2009.02.00219236958
|
|
35.
|
PA CeruttiProoxidant states and tumor
promotionScience227375381198510.1126/science.29814332981433
|
|
36.
|
AH SpragueRA KhalilInflammatory cytokines
in vascular dysfunction and vascular diseaseBiochem
Pharmacol78539552200910.1016/j.bcp.2009.04.02919413999
|
|
37.
|
P QinX TangMM EllosoDC HarnishBile acids
induce adhesion molecule expression in endothelial cells through
activation of reactive oxygen species, NF-kappaB, and p38Am J
Physiol Heart Circ
Physiol291H741H747200610.1152/ajpheart.01182.200516582018
|
|
38.
|
R SchreckB MeierDN MannelW DrogePA
BaeuerleDithiocarbamates as potent inhibitors of nuclear factor
kappa B activation in intact cellsJ Exp
Med17511811194199210.1084/jem.175.5.11811314883
|
|
39.
|
JM LuPH LinQ YaoC ChenChemical and
molecular mechanisms of antioxidants: experimental approaches and
model systemsJ Cell Mol
Med14840860201010.1111/j.1582-4934.2009.00897.x19754673
|