Introduction
AMP-activated protein kinase (AMPK) is generally
known to regulate multiple metabolic pathways (1). AMPK has been identified as a
mammalian protein kinase that is allosterically activated by AMP
and is able to phosphorylate and inactivate enzymes of lipid
synthesis (1). It is currently
recognized that AMPK is a key sensing enzyme involved in the
regulation of cellular energy homeostasis (2–4).
AMPK is activated in mammalian cells by a variety of physiological
and pathological stresses that increase the intracellular AMP:ATP
ratio, either by increasing ATP consumption or by decreasing ATP
production. Activated AMPK acts to restore cellular energy balance
by ATP-generating pathways such as fatty acid oxidation, while
simultaneously inhibiting ATP-utilizing pathways. It is well
recognized that bone metabolism is regulated mainly by two
functional cells, osteoblasts and osteoclasts (5). The former cells are responsible for
bone formation and the latter cells are responsible for bone
resorption. These functional cells are closely coordinated via
humoral factors or by direct cell-to-cell interaction (5,6).
Regarding AMPK in bone metabolism, it has been shown that activated
AMPK regulates bone formation and bone mass in vitro
(7,8). In osteoblasts, activation of AMPK
reportedly stimulates their differentiation and inhibits apoptosis
(9,10). We previously showed that AMPK
plays a role in vascular endothelial growth factor synthesis in
osteoblast-like MC3T3-E1 cells (11). However, the exact role of AMPK in
bone metabolism has not yet been elucidated.
It is well known that interleukin-6 (IL-6) is a
multifunctional cytokine that has crucial effects on a variety of
cell functions such as promoting B-cell differentiation, T-cell
activation and inducing acute phase proteins (6,12,13). With regard to bone metabolism,
IL-6 has been shown to promote osteoclast formation and stimulate
bone resorption (6,13–15). It has been reported that potent
bone resorptive agents such as tumor necrosis factor-α, IL-1 and
prostaglandin (PG) E2 stimulate IL-6 synthesis in
osteoblasts (14,16,17). Evidence suggests that IL-6, which
is synthesized and secreted from osteoblasts, plays an important
role as a downstream effector of bone resorptive agents in bone
metabolism. It is well known that PGs act as autocrine/paracrine
modulators of osteoblasts (18).
Among them, prostaglandin F2α (PGF2α) is
recognized to be a potent bone resorptive agent in bone metabolism.
It has been reported that PGF2α stimulates the
proliferation of osteoblasts and inhibits the differentiation
(18). We previously reported
that PGF2α stimulates the IL-6 synthesis in
osteoblast-like MC3T3-E1 cells (19) and that p44/p42 mitogen-activated
protein (MAP) kinase and p38 MAP kinase among the MAP kinase
superfamily play a role in PGF2α-induced IL-6 synthesis
in these cells (20,21). However, the detailed mechanism
behind the PGF2α-stimulated IL-6 synthesis in
osteoblasts remains to be clarified.
In the present study, we investigated the
involvement of AMPK in PGF2α-stimulated IL-6 synthesis
in osteoblast-like MC3T3-E1 cells. We herein showed that AMPK
positively regulates PGF2α-stimulated IL-6 synthesis at
a point upstream from p38 MAP kinase activation in these cells.
Materials and methods
Materials
PGF2α and mouse IL-6 enzyme-linked
immunosorbent assay (ELISA) kit were purchased from R&D
Systems, Inc. (Minneapolis, MN). Compound C was obtained from
Calbiochem-Novabiochem Co. (La Jolla, CA). Normal human osteoblasts
(NHOst) were purchased from Cambrex (Charles City, IA).
Phospho-specific AMPK α-subunit (Thr-172), phospho-specific AMPK
α-subunit (Ser-485), AMPK α-subunit, phospho-specific AMPK
β-subunit (Ser-108), phospho-specific AMPK β-subunit (Ser-182),
AMPK β-subunit, phospho-specific acetyl-CoA carboxylase,
phospho-specific p44/p42 MAP kinase, p44/p42 MAP kinase,
phospho-specific p38 MAP kinase and p38 MAP kinase antibodies were
purchased from Cell Signaling Technology, Inc. (Beverly, MA). The
GAPDH antibody was obtained from Santa Cruz Biotechnology, Inc.
(Santa Cruz, CA). An ECL western blotting detection system was
purchased from GE Healthcare UK, Ltd. (Buckinghamshire, UK).
Control short interfering RNA (siRNA) (Silencer®
Negative Control #1 siRNA) was purchased from Ambion (Austin, TX).
AMPK α1-subunit siRNA (SI0 1388219) and the OmniScript Reverse
Transcriptase kit were purchased from Qiagen GmbH (Hilden,
Germany). siLentFect™ was purchased from Bio-Rad (Hercules, CA).
TRIzol reagent was purchased from Invitrogen (Carlsbad, CA). Fast
Start DNA Master SYBR-Green I was purchased from Roche Diagnostics
(Mannheim, Germany). Other materials and chemicals were obtained
from commercial sources.
Cell culture
Cloned osteoblast-like MC3T3-E1 cells that were
derived from newborn mouse calvaria (22) were maintained as previously
described (23). Briefly, the
cells were cultured in α-minimum essential medium (α-MEM)
containing 10% fetal calf serum (FCS) at 37°C in a humidified
atmosphere of 5% CO2/95% air. The cells were seeded into
35-mm (5×104) or 90-mm (2×105) diameter
dishes in α-MEM containing 10% FCS. After 5 days, the medium was
replaced with α-MEM containing 0.3% FCS. The cells were used for
experiments after 48 h.
NHOst were seeded into 35-mm (5×104)
diameter dishes in α-MEM containing 10% FCS. After 6 days, the
medium was replaced with α-MEM containing 0.3% FCS. The cells were
used for experiments after 48 h.
IL-6 assay
The cultured cells were stimulated with 10 μM
PGF2α in 1 ml of α-MEM containing 0.3% FCS for the
indicated periods. When indicated, the cells were pretreated with
various doses of compound C for 60 min. The conditioned medium was
collected at the end of the incubation and the IL-6 concentration
was measured by the IL-6 ELISA kit.
siRNA transfection
To knock down the AMPK α-subunit in MC3T3-E1 cells,
the cells were transfected with the control siRNA or the AMPK
α1-subunit siRNA using siLentFect according to the manufacturer’s
protocol. In brief, the cells (1×105) were seeded in a
35-mm diameter dish in α-MEM containing 10% FCS and subcultured for
48 h. The cells were then incubated with 50 nM siRNA-siLentFect
complexes. After 24 h, the medium was replaced with α-MEM
containing 0.3% FCS. The cells were used for experiments after 24
h.
Western blot analysis
Western blot analysis was performed as previously
described (24). The cultured
cells were stimulated with PGF2α or vehicle in α-MEM
containing 0.3% FCS for the indicated periods. When indicated, the
cells were pretreated with various doses of compound C for 60 min.
The cells were washed twice with phosphate-buffered saline and then
lysed, homogenized and sonicated in a lysis buffer containing 62.5
mM Tris/HCl (pH 6.8), 3% sodium dodecyl sulfate (SDS), 50 mM
dithiothreitol and 10% glycerol. The cytosolic fraction was
collected as a supernatant after centrifugation at 125,000 × g for
10 min at 4°C. SDS-polyacrylamide gel electrophoresis (PAGE) was
performed according to Laemmli et al (25) on 10% polyacrylamide gel. The
protein (20 μg) was fractionated and transferred onto an
Immun-Blot® PVDF Membrane (Bio-Rad, Hercules, CA).
Membranes were blocked with 5% fat-free dry milk in Tris-buffered
saline-Tween [TBS-T; 20 mM Tris/HCl (pH 7.6), 137 mM NaCl, 0.1%
Tween-20] for 2 h before incubation with the primary antibodies.
Phospho-specific AMPK α-subunit (Thr-172), phospho-specific AMPK
α-subunit (Ser-485), AMPK α-subunit, phospho-specific AMPK
β-subunit (Ser-108), phospho-specific AMPK β-subunit (Ser-182),
AMPK β-subunit, phospho-specific acetyl-CoA carboxylase,
phospho-specific p44/p42 MAP kinase, p44/p42 MAP kinase,
phospho-specific p38 MAP kinase, p38 MAP kinase, phospho-specific
SAPK/JNK and SAPK/JNK antibodies or GAPDH antibody were used as
primary antibodies. Peroxidase-labeled antibody raised in goat
against rabbit IgG (KPL, Inc., Gaithersburg, MD) was used as the
secondary antibody. The primary and secondary antibodies were
diluted at 1:1,000 with 5% fat-free dry milk in TBS-T. Peroxidase
activity on the membrane was visualized on X-ray film by means of
the ECL western blotting detection system.
Real-time RT-PCR
The cultured cells were stimulated with 10 μM
PGF2α for the indicated periods. Total RNA was isolated
and transcribed into cDNA using TRIzol reagent and the OmniScript
Reverse Transcriptase kit. Real-time RT-PCR was performed using a
Light Cycler system (Roche Diagnostics, Basel, Switzerland) in the
capillaries and Fast Start DNA Master SYBR-Green I provided with
the kit. Sense and antisense primers for mouse IL-6 and GAPDH mRNA
were purchased from Takara Bio, Inc. (Tokyo, Japan) (primer set ID
MA039013). The amplified products were determined by a melting
curve analysis and agarose electrophoresis. IL-6 mRNA levels were
normalized with those of GAPDH mRNA.
Determination
The absorbance of enzyme immunoassay samples was
measured at 450 nm with an EL 340 Bio Kinetic Reader (Bio-Tek
Instruments, Inc., Winooski, VT).
Statistical analysis
Data were analyzed by ANOVA followed by the
Bonferroni method for multiple comparisons between pairs and a
P<0.05 was considered to indicate a statistically significant
difference. All data are presented as the means ± SEM of triplicate
independent determinations.
Results
Effect of PGF2α on the
phosphorylation of AMP-activated protein kinase in MC3T3-E1
cells
It is generally known that the phosphorylation of
AMPK is essential for its activation (26). Therefore, we first examined the
effect of PGF2α on the phosphorylation of AMPK in
osteoblast-like MC3T3-E1 cells in order to clarify whether
PGF2α induces the activation of AMPK in osteoblasts.
PGF2α markedly stimulated the phosphorylation of the
AMPK α-subunit (Thr-172) (Fig.
1). In contrast, levels of phosphorylated AMPK β-subunit
(Ser-108) or phosphorylated AMPK β-subunit (Ser-182) were barely
affected by PGF2α (Fig.
1). The effect of PGF2α on the phosphorylation of
AMPK α-subunit (Thr-172) reached its peak within 2 min and
thereafter decreased.
Effect of compound C on
PGF2α-stimulated IL-6 release in MC3T3-E1 cells
In our previous study (19), we showed that PGF2α
stimulates IL-6 synthesis in osteoblast-like MC3T3-E1 cells. In
order to investigate whether AMPK plays a role in
PGF2α-induced synthesis of IL-6 in MC3T3-E1 cells, we
examined the effect of compound C, an inhibitor of AMPK (27), on the PGF2α-stimulated
release of IL-6. Compound C significantly reduced
PGF2α-stimulated IL-6 release (Fig. 2). The suppressive effect of
compound C on the IL-6 release was dose-dependent in the range
between 1 and 20 μM. Twenty micromoles of compound C caused
an ∼55% inhibition in the PGF2α effect.
Effects of PGF2α on the
phosphorylation of acetyl-CoA carboxylase in MC3T3-E1 cells
Acetyl-CoA carboxylase, which regulates lipid
synthesis, is a direct substrate of AMPK (1). PGF2α markedly induced the
phosphorylation of acetyl-CoA carboxylase in MC3T3-E1 cells
(Fig. 3). The effect of
PGF2α on acetyl-CoA carboxylase phosphorylation reached
a peak within 6 min and thereafter decreased. Compound C reduced
the PGF2α-induced phosphorylation level of acetyl-CoA
carboxylase in a dose-dependent manner (Fig. 4).
Effect of compound C on the
PGF2α-stimulated IL-6 release in human osteoblasts
In addition, we examined the effect of compound C
(27), on
PGF2α-induced release of IL-6 in another type of
osteoblast, NHOst, a human osteoblastic cell type. We found that
PGF2α markedly stimulated IL-6 release in NHOst.
Compound C significantly suppressed the PGF2α-induced
release of IL-6 (Fig. 5).
Compound C (1 μM) caused an ∼45% inhibition in the
PGF2α effect.
Effect of compound C on the
PGF2α-stimulated IL-6 mRNA expression in MC3T3-E1
cells
In order to clarify whether the suppression of
PGF2α-stimulated IL-6 synthesis by compound C is
mediated via a transcriptional event, we examined the effect of
compound C on PGF2α-induced expression level of IL-6
mRNA in MC3T3-E1 cells. Compound C, which alone had a slight
suppressive effect on the basal IL-6 mRNA expression level,
significantly reduced the PGF2α-induced level of IL-6
mRNA (Fig. 6).
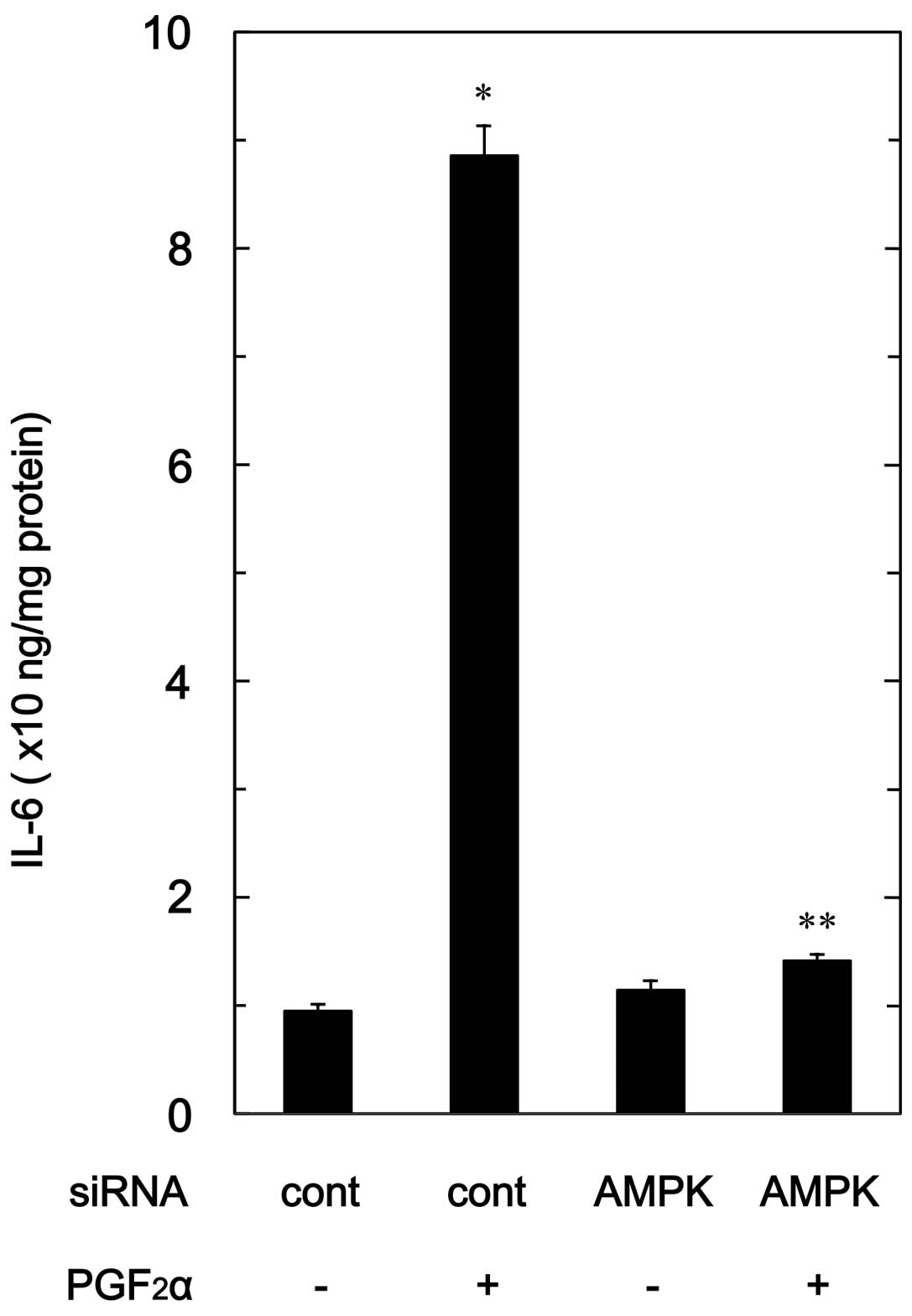
Effect of AMPK siRNA on
PGF2α-stimulated IL-6 release in MC3T3-E1 cells
To confirm the findings from the compound C
experimental series in osteoblast-like MC3T3-E1 cells, we examined
the effect of AMPK downregulation by siRNA on the
PGF2α-stimulated IL-6 release. PGF2α-induced
levels of IL-6 in AMPK α1-subunit-downregulated cells were markedly
suppressed compared with levels in the control siRNA-transfected
cells (Fig. 7).
Effects of compound C on
PGF2α-induced phosphorylation of p44/p42 MAP kinase or
p38 MAP kinase in MC3T3-E1 cells
The MAP kinase superfamily such as p44/p42 MAP
kinase, p38 MAP kinase and stress-activated protein
kinase/c-Jun N-terminal kinase (SAPK/JNK) plays a central
role in the transduction of the various messages of a variety of
agonists (28). In our previous
studies (20,21), we demonstrated that
PGF2α induces the activation of p44/p42 MAP kinase, p38
MAP kinase and SAPK/JNK in osteoblast-like MC3T3-E1 cells and that
p44/p42 MAP kinase and p38 MAP kinase but not SAPK/JNK function as
positive regulators in PGF2α-stimulated IL-6 synthesis.
In order to elucidate whether the AMPK effect on
PGF2α-stimulated IL-6 synthesis is due to the activation
of these MAP kinases in MC3T3-E1 cells, we examined the effect of
compound C on PGF2α-induced phosphorylation of p44/p42
MAP kinase or p38 MAP kinase. PGF2α-induced
phosphorylation of p44/p42 MAP kinase was not suppressed by
compound C up to 30 μM (Fig.
8). On the contrary, compound C markedly attenuated the
PGF2α-induced phosphorylation level of p38 MAP kinase in
a dose-dependent manner in the range between 3 and 30 μM
(Fig. 9).
Discussion
In the present study, we showed that
PGF2α markedly stimulated the phosphorylation of
AMP-activated protein kinase (AMPK) α-subunit (Thr-172) among the
phosphorylation sites of AMPK in osteoblast-like MC3T3-E1 cells. It
is generally recognized that AMPK plays a crucial role as a central
regulator of cellular energy homeostasis (2). AMPK is a heterotrimeric complex that
consists of three subunits, α, β and γ (26,29). Among the three subunits of AMPK,
the α-subunit is recognized to be a catalytic site whereas β- and
γ-subunits are considered as regulatory sites. It has been reported
that the phosphorylation of the AMPK α-subunit at Thr-172 is
essential for enzyme activation (26). We also showed that
PGF2α induced acetyl-CoA carboxylase phosphorylation in
MC3T3-E1 cells. Acetyl-CoA carboxylase is a downstream target of
AMPK, and phosphorylated acetyl-CoA carboxylase inhibits the
synthesis of lipids such as fatty acid and cholesterol (1,26).
We demonstrate that the time course of PGF2α-induced
phosphorylation of the AMPK α-subunit at Thr-172 was more rapid
than that of acetyl-CoA carboxylase (Figs. 1 and 3). In addition, we showed that compound
C, an inhibitor of AMPK (27),
reduced the levels of acetyl-CoA carboxylase phosphorylation
induced by PGF2α. Based on our findings, it is most
likely that PGF2α stimulates the activation of AMPK in
osteoblast-like MC3T3-E1 cells.
We next investigated whether AMPK activation plays a
role in PGF2α-stimulated IL-6 synthesis in
osteoblast-like MC3T3-E1 cells. Compound C significantly inhibited
the PGF2α-stimulated release of IL-6 in a dose-dependent
manner. These findings suggest that the AMPK activated by
PGF2α is involved in IL-6 synthesis in MC3T3-E1 cells.
In addition, we showed that compound C markedly inhibited
PGF2α-induced IL-6 mRNA expression. It is likely that
AMPK regulates the synthesis of IL-6. Additionally, we found that
compound C suppressed the IL-6 levels stimulated by
PGF2α in NHOst, a human osteoblast cell type. Thus, it
is probable that the effect of compound C on
PGF2α-induced IL-6 synthesis is common in mammalian
osteoblasts. Furthermore, we demonstrated that downregulation of
AMPK by siRNA markedly reduced the PGF2α-stimulated IL-6
release in MC3T3-E1 cells. Taking our results into account, it is
likely that PGF2α stimulates the activation of AMPK in
osteoblast-like MC3T3-E1 cells and that AMPK positively regulates
PGF2α-induced IL-6 synthesis.
In our previous studies (20,21), we reported that p44/p42 MAP kinase
and p38 MAP kinase but not SAPK/JNK among the MAP kinase
superfamily act as positive regulators in
PGF2α-stimulated IL-6 synthesis in osteoblast-like
MC3T3-E1 cells. Therefore, we investigated the relationship between
AMPK and MAP kinase signaling, p44/p42 MAP kinase or p38 MAP kinase
in IL-6 synthesis. Compound C markedly suppressed
PGF2α-induced phosphorylation levels of p38 MAP kinase
without affecting the levels of p44/p42 MAP kinase phosphorylation.
These findings suggest that AMPK functions in the
PGF2α-stimulated activation of p38 MAP kinase in
osteoblast-like MC3T3-E1 cells. In the present study, we showed
that the maximum effect of PGF2α on the phosphorylation
of the AMPK α-subunit (Thr-172) was observed within 2 min after
stimulation. In contrast, in our previous study (30), we showed that the phosphorylation
of p38 MAP kinase reached a peak at 10 min after the stimulation of
PGF2α. The time course of PGF2α-induced
phosphorylation of AMPK appears to be more rapid than that of p38
MAP kinase. Therefore, it is reasonable that PGF2α
induces the activation of p38 MAP kinase via AMPK in MC3T3-E1
cells. Collectively, PGF2α stimulates IL-6 synthesis via
AMPK, which plays a role at least partly through the regulation of
p38 MAP kinase in osteoblast-like MC3T3-E1 cells.
AMPK is a central regulator in cellular energy
homeostasis such as lipid synthesis (1,31).
With regard to bone metabolism, accumulating evidence suggests that
AMPK stimulates bone formation and mineralization, resulting in an
increase in bone mass (7,8). As for osteoblast function, it has
recently been shown that the activation of AMPK inhibits
palmitate-induced apoptosis in osteoblasts (9). AMPK was reported to stimulate
osteoblast differentiation via induction of Runx2 expression
(10). In addition, it has
recently been shown that inhibition of AMPK suppresses
osteoprotegerin secretion in osteoblasts (32). Based on our present findings, it
is possible that inhibition of AMPK reduces the
PGF2α-stimulated IL-6 synthesis in osteoblasts.
Osteoporosis is a major clinical problem in advanced countries. The
pathology of osteoporosis involves the reduction in bone mineral
density, resulting in susceptibility to bone fracture (33). IL-6 synthesized by osteoblasts is
generally recognized as a potent bone resorptive agent and induces
osteoclast formation (6,14). Therefore, we speculate that
PGF2α-activated AMPK in osteoblasts is a potent
modulator in bone metabolism via the fine-tuning of the local
cytokine network, such as synthesis of IL-6. The appropriate
regulation of AMPK in osteoblasts may be a potential molecular
therapeutic strategy for bone metabolic disease such as
osteoporosis. However, the detailed mechanisms of AMPK in bone
metabolism are not precisely known. Further investigation is
necessary to clarify the exact roles of AMPK in osteoblasts.
Taken together, our results revealed that an AMPK
inhibitor decreases PGF2α-stimulated IL-6 synthesis via
suppression of p38 MAP kinase in osteoblasts.
Acknowledgements
We are grateful to Yoko Kawamura for
her skillful technical assistance. This investigation was supported
in part by a Grant-in-Aid for Scientific Research (16590873 and
16591482) for the Ministry of Education, Science, Sports and
Culture of Japan, the Foundation for Growth Science and the
Research Funding for Longevity Sciences (21-4, 22-4 and 23-9) from
the National Center for Geriatrics and Gerontology (NCGG),
Japan.
References
|
1.
|
S FogartyDG HardieDevelopment of protein
kinase activators: AMPK as a target in metabolic disorders and
cancerBiochim Biophys
Acta1804581591201010.1016/j.bbapap.2009.09.01219778642
|
|
2.
|
DG HardieSA HawleyJW ScottAMP-activated
protein kinase - development of the energy sensor conceptJ
Physiol574715200610.1113/jphysiol.2006.10894416644800
|
|
3.
|
R LageC DieguezA Vidal-PuigM LopezAMPK: a
metabolic gauge regulating whole-body energy homeostasisTrends Mol
Med14539549200810.1016/j.molmed.2008.09.00718977694
|
|
4.
|
GR SteinbergBE KempAMPK in health and
diseasePhysiol
Rev8910251078200910.1152/physrev.00011.200819584320
|
|
5.
|
G KarsentyEF WagnerReaching a genetic and
molecular understanding of skeletal developmentDev
Cell2389406200210.1016/S1534-5807(02)00157-011970890
|
|
6.
|
S Kwan TatM PadrinesS TheoleyreD HeymannY
FortunIL-6, RANKL, TNF-alpha/IL-1: interrelations in bone
resorption pathophysiologyCytokine Growth Factor Rev1549602004
|
|
7.
|
M ShahB KolaA BataveljicTR ArnettB
ViolletL SaxonM KorbonitsC ChenuAMP-activated protein kinase (AMPK)
activation regulates in vitro bone formation and bone
massBone47309319201010.1016/j.bone.2010.04.59620399918
|
|
8.
|
I KanazawaT YamaguchiS YanoM YamauchiT
SugimotoMetformin enhances the differentiation and mineralization
of osteoblastic MC3T3-E1 cells via AMPK activation as well as eNOS
and BMP-2 expressionBiochem Biophys Res
Commun375414419200810.1016/j.bbrc.2008.08.03418721796
|
|
9.
|
JE KimMW AhnSH BaekIK LeeYW KimJY KimJM
DanSY ParkAMPK activator, AICAR, inhibits palmitate-induced
apoptosis in
osteoblastBone43394404200810.1016/j.bone.2008.03.02118502715
|
|
10.
|
WG JangEJ KimKN LeeHJ SonJT
KohAMP-activated protein kinase (AMPK) positively regulates
osteoblast differentiation via induction of Dlx5-dependent Runx2
expression in MC3T3E1 cellsBiochem Biophys Res
Commun40410041009201110.1016/j.bbrc.2010.12.09921187071
|
|
11.
|
K KatoH TokudaS AdachiR
Matsushima-NishiwakiH NatsumeK YamakawaY GuT OtsukaO
KozawaAMP-activated protein kinase positively regulates
FGF-2-stimulated VEGF synthesis in osteoblastsBiochem Biophys Res
Commun400123127201010.1016/j.bbrc.2010.08.02420708602
|
|
12.
|
S AkiraT TagaT KishimotoInterleukin-6 in
biology and medicineAdv
Immunol54178199310.1016/S0065-2776(08)60532-5
|
|
13.
|
D HeymannAV Roussellegp130 Cytokine family
and bone
cellsCytokine1214551468200010.1006/cyto.2000.074711023660
|
|
14.
|
Y IshimiC MiyauraCH JinT AkatsuF AbeY
NakamuraY YamaguchiS YoshikiT MatsudaT HiranoT KishimotoT SudaIL-6
is produced by osteoblasts and induces bone resorptionJ
Immunol1453297330319902121824
|
|
15.
|
GD RoodmanInterleukin-6: an osteotropic
factor?J Bone Miner
Res7475478199210.1002/jbmr.56500705021615755
|
|
16.
|
AJ LittlewoodJ RussellGR HarveyDE
HughesRGG RusselM GowenThe modulation of the expression of IL-6 and
its receptor in human osteoblasts in
vitroEndocrinology12915131520199110.1210/endo-129-3-15131714833
|
|
17.
|
M HelleJPJ BrakenhoffER De GrootLA
AardenInterleukin 6 is involved in interleukin 1-induced
activitiesEur J Immunol18957959199810.1002/eji.1830180619
|
|
18.
|
CC PilbeamJR HarrisonLG
RaiszProstaglandins and bone metabolismPrinciples of Bone BiologyJP
BilezikianLG RaiszGA RodanAcademic PressSan Diego,
CA979994200210.1016/B978-012098652-1/50156-6
|
|
19.
|
O KozawaA SuzukiH TokudaT
UematsuProstaglandin F2α stimulates interleukin-6
synthesis via activation of PKC in osteoblast-like cellsAm J
Physiol272E208E21119979124324
|
|
20.
|
H TokudaO KozawaA HaradaT Uematsup42/p44
mitogen-activated protein kinase activation is involved in
prostaglandin F2α-induced interleukin-6 synthesis in
osteoblastsCell
Signal11325330199910.1016/S0898-6568(98)00048-510376804
|
|
21.
|
C MinamitaniT OtsukaS TakaiR
Matsushima-NishiwakiS AdachiY HanaiJ MizutaniH TokudaO
KozawaInvolvement of Rho-kinase in prostaglandin
F2α-stimulated interleukin-6 synthesis via p38
mitogen-activated protein kinase in osteoblastsMol Cell
Endocrinol2912732200818586382
|
|
22.
|
H SudoH KodamaY AmagaiS YamamotoS KasaiIn
vitro differentiation and calcification in a new clonalosteogenic
cell line derived from newborn mouse calvariaJ Cell
Biol96191198198310.1083/jcb.96.1.1916826647
|
|
23.
|
O KozawaH TokudaM MiwaJ KotoyoriY
OisoCross-talk regulation between cyclic AMP production and
phosphoinositide hydrolysis induced by prostaglandin E2
in osteoblast-like cellsExp Cell
Res198130134199210.1016/0014-4827(92)90158-51309194
|
|
24.
|
K KatoH ItoK HasegawaY InagumaO KozawaT
AsanoModulation of the stress-induced synthesis of hsp27 and α
B-crystallin by cyclic AMP in C6 rat glioma cellsJ
Neurochem669469501996
|
|
25.
|
UK LaemmliCleavage of structural proteins
during the assembly of the head of bacteriophage
T4Nature227680685197010.1038/227680a05432063
|
|
26.
|
SA HawleyM DavisonA WoodsSP DaviesRK BeriD
CarlingDG HardieCharacterization of the AMP-activated protein
kinase kinase from rat liver and identification of threonine 172 as
the major site at which it phosphorylates AMP-activated protein
kinaseJ Biol
Chem2712787927887199610.1074/jbc.271.44.278798910387
|
|
27.
|
G ZhouR MyersY LiY ChenX ShenJ
Fenyk-MelodyM WuJ VentreT DoebberN FujiiN MusiMF HirshmanLJ
GoodyearDE MollerRole of AMP-activated protein kinase in mechanism
of metformin activationJ Clin
Invest10811671174200110.1172/JCI1350511602624
|
|
28.
|
JM KyriakisJ AvruchMammalian
mitogen-activated protein kinase signal transduction pathways
activated by stress and inflammationPhysiol
Rev81807869200111274345
|
|
29.
|
D StapletonG GaoBJ MichellJ WidmerK
MitchelhillT TehCM HouseLA WittersBE KempMammalian 5′-AMP-activated
protein kinase non-catalytic subunits are homologs of proteins that
interact with yeast Snf1 protein kinaseJ Biol
Chem26929343293461994
|
|
30.
|
H TokudaS TakaiR Matsushima-NishiwakiS
AkamatsuY HanaiT HosoiA HaradaT OhtaO Kozawa(−)-Epigallocatechin
gallate enhances prostaglandin F2α-induced VEGF
synthesis via up-regulating SAPK/JNK activation in osteoblastsJ
Cell Biochem100114611532007
|
|
31.
|
GA RutterI LeclercThe AMP-regulated kinase
family: enigmatic targets for diabetes therapyMol Cell
Endocrinol2974149200910.1016/j.mce.2008.05.02018611432
|
|
32.
|
Q MaiZ ZhangS XuM LuR ZhouL ZhaoC JiaZ
WenD JinX BaiMetformin stimulates osteoprotegerin and reduces RANKL
expression in osteoblasts and ovariectomized ratsJ Cell
Biochem11229022909201110.1002/jcb.2320621618594
|
|
33.
|
A UnnanuntanaBP GladnickE DonnellyJM
LaneThe assessment of fracture riskJ Bone Joint Surg
Am92743753201010.2106/JBJS.I.00919
|