Introduction
Epstein-Barr virus (EBV) has been identified as a
DNA virus that is associated with human malignancies, including
Burkitt’s lymphoma, Hodgkin’s disease and nasopharyngeal carcinoma
(NPC) (1,2). Latent membrane protein 1 (LMP1) is
an important oncogenic protein among EBV-encoded proteins and is
expressed in up to 90% of NPC patients. It thus plays an important
role in the development of NPC (3,4).
Previous work has shown that LMP1 is involved in multiple NPC
biological processes including cell proliferation, apoptosis,
invasion and metastasis, by inducing the activation of NF-κB,
JAK/STAT, PKC and the MAPK signaling pathways (5) and their downstream genes including
p53 and survivin (6,7).
The tumor suppressor gene p53 is a critical
mediator of cell cycle, DNA repair, cell differentiation and
apoptosis. Many human tumors are associated with p53 mutations,
supporting its pivotal role as a key tumor suppressor in
tumorigenesis. Unlike in most human tumors, p53 accumulates in NPC
and the mutation rate of p53 is <10% (8,9).
Immunohistochemical analysis of NPC biopsies indicates that p53
accumulation is significantly associated with LMP1 expression
(10,11). In our previous study, LMP1 was
found to increase the transcriptional activity and expression of
both wild-type and mutant p53 in NPC (6,12).
LMP1 could mediate p53 phosphorylation at Ser15, Ser20, Ser392 and
Thr81, indicating potential functional activity of p53 in NPC
progression (13). Although other
reports have also shown a possible role of p53 in NPC (14,15), the biological function and
potential downstream target of accumulated p53 in NPC still remains
unclear.
Survivin, a member of the inhibitor of apoptosis
(IAP) family identified in 1997 (16), is widely expressed in fetal
tissues and most tumor tissues. Survivin is overexpressed in
multiple tumors, and corresponds with poor prognosis (17). Survivin is highly expressed at the
G2/M phase in a cell cycle-regulated manner, and also promotes G1/S
cell cycle progression by translocating into the nucleus and
forming a complex with CDK4 (18). As a mitotic substrate of
Cdc2/cyclin B1, survivin can be phosphorylated on Thr34 on the
mitotic apparatus, which contributes to the regulation of cell
division through an interaction with caspase-9 (19). Thus, survivin has dual functions
in cell cycle regulation and apoptosis inhibition. We previously
showed that LMP1 could increase the activity of survivin through
the NF-κB and AP-1 signaling pathways in NPC (7,19).
Remarkably, the survivin promoter contains a p53 binding
element located in the survivin 230-bp basic core promoter
region (20,21), indicating the possible regulation
of survivin by LMP1 via p53 in NPC.
Here, using siRNA technology to knockdown the
expression of p53, we found that LMP1 upregulated survivin protein
expression and its phosphorylation by p53 due to the
transactivation of the survivin promoter. LMP1 caused the
translocation of p53 into the nucleus with survivin, suggesting
that survivin is the key downstream target of p53. We further found
that accumulated p53 by LMP1 promoted G1/S cell cycle progression,
but did not induce apoptosis in NPC pathogenesis.
Materials and methods
Cell culture and plasmids
CNE1 cells comprise an LMP1-negative, highly
differentiated nasopharyngeal carcinoma cell line with mutant p53
(8). CNE1-LMP1 NPC cells are a
stably transfected cell line, which was established by introducing
LMP1 cDNA into CNE1 NPC cells. CNE1 and CNE1-LMP1 NPC cells
were maintained in RPMI-1640 medium supplemented with 10% fetal
calf serum. NP69-pLNSX and NP69-LMP1 cells are SV40-transformed,
immortalized normal nasopharyngeal cell lines (33), with no p53 mutation (12). These cells were cultured in
defined keratinocyte serum-free medium (KSFM) (Gibco Life
Technologies, Basel, Switzerland) supplemented with growth factors.
All cell lines were maintained at 37°C with 5% CO2. The
pSUPER-p53siRNA is a plasmid with an siRNA targeting p53 inserted
in the pSUPER vector. This vector was a generous gift from
Professor Qiao Wu (Key Laboratory of Ministry of Education for Cell
Biology and Tumor Cell Engineering, Xiamen University, China). The
pGL3-Sur1.8kb (pGL3.basic.survivin.promoter1.8kb) is a
survivin promoter-luciferase reporter construct obtained
from Professor Ningzhi Xu (Cancer Research Institute, Chinese
Academy of Medical Sciences). The pSV-β-galactosidase control
vector was purchased from Promega (Southampton, UK).
Western blot analyses
Cells were collected and washed with ice-cold PBS 3
times. Lysis buffer (50 mM Tris-HCl, 1 mM EDTA, 20 g/l SDS, 5 mM
DTT and 10 mM PMSF) was added and cells were left on ice for 30
min. Cells were then boiled for 10 min followed by ultrasonication
for 30 sec. All procedures were carried out at 4°C. Proteins were
collected by centrifugation at 10,000 × g for 10 min. Protein
concentrations were determined using the BCA protein assay reagent
(Pierce Chemical Co., Rockford, IL), with bovine serum albumin as a
standard. For western blot analysis, 50 μg of total protein was
loaded onto an 8–12% Tris-glycine polyacrylamide gel and subjected
to electrophoresis. Proteins were visualized by ECL
chemiluminescence reagents (Pierce Chemical Co.). Primary
antibodies specific for human p53 (sc-126), phosphorylated p53
(Ser20) (sc-18079), survivin (sc-8807), phosphorylated survivin
(Thr34) (sc-23758), caspase-3 (sc-7272), CDK2 (sc-163), CDK4
(sc-260), cyclin D1 (sc-20044), α-tubulin (sc-5286), nucleolin
(sc-8031) and secondary antibodies for goat anti-rabbit IgG-HRP
(sc-2004) and goat anti-mouse IgG-HRP (sc-2005) were all purchased
from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA).
Quantitative real-time PCR
Total-RNA was isolated using the TRIzol (Invitrogen,
Carlsbad, CA, USA) reagent following the manufacturer’s suggested
protocols. Reverse transcription PCR was performed by using the
Reverse Transcription System (Promega, Madison, WI). Each 25 μl of
PCR reaction mixture was prepared using the SYBR® Premix
Ex Taq™ kit (Takara Bio, Inc.). The primer sequences for
survivin were as follows: forward, 5′-AGGTGCCTGTTGAATCTG-3′
and reverse, 5′-GACGCTTCCTATCACTCTATT-3′; the β-actin primer
sequences were forward, 5′-TTCCAGCCTTCCTTCCTGGG-3′ and reverse,
5′-TTGCGCTCAGGAGGAGCAAT-3′. The amplification conditions were set
up according to the protocol included with the SYBR Premix Ex Taq™
kit. Samples were run in triplicate for each experiment using the
7500 Real-Time PCR System (Applied Biosystems, Foster City, CA).
The relative amount of mRNA was calculated using the comparative CT
method after normalization to β-actin mRNA levels.
Luciferase assays
Survivin promoter-luciferase reporter
constructs (pGL3-Sur1.8kb) in combination with the
pSV-β-galactosidase control construct were transfected into cells
48 h after pSUPER-p53-siRNA transfection, using Lipofectamine™
2000, according to protocols provided by the manufacturer
(Invitrogen). After 24 h, cells were harvested and lysed with
reporter lysis buffer (RLB; Promega) and the luciferase activity
was determined using the Luciferase Reporter Assay System (Promega)
according to the manufacturer’s instructions. Experiments were
performed in quadruplicate, and the statistical significance was
assessed by a paired t-test.
Electrophoretic gel mobility shift assay
(EMSA)
Nuclear extracts were prepared by the use of the
NE-PER Nuclear and Cytoplasmic Extraction kit (Pierce) in
accordance with the manufacturer’s protocol. The p53
oligonucleotide probes were synthesized (Invitrogen). The sequences
were as follows: (Bio)-p53, sense, 5′-(Biotin)-GCCTAAGAGGGCGTGCGCTC
CCGACATGCCCCGCGG-3′ and antisense, 5′-(Biotin)-CC
GCGGGGCATGTCGGGAGCGCACGCCCTCTTAGGC-3′; NS-p53, sense,
5′-CAGGGACGATATGGATAGATTTC GCTGGGT-3′ and antisense,
5′-ACCCAGCGAAATCTATCC ATATCGTCCCTG-3′; (Bio)-mut-p53, sense,
5′-(Biotin)-GC CTAAGAGGTCTCTCGCTCCCGAAAGACCCCGCGG-3′ and antisense,
5′-Biotin-CCGCGGGGTCTTTCGGGAGC GAGAGACCTCTTAGGC-3′. EMSAs were
performed using protocols provided in the LightShift™
Chemiluminescent EMSA kit (Pierce). Labeled (2 μl) oligonucleotides
and nucleoproteins (10 μg) were mixed in binding buffer (Pierce)
and incubated for 15 min at room temperature. Samples were
subjected to electrophoresis in 5% non-denaturing polyacrylamide
gel and transferred to a Biodyne™ B Nylon membrane (Pierce). The
protein bands were detected using ECL chemiluminescence reagents
(Pierce).
Chromatin immunoprecipitation (ChIP)
assay
ChIP assays were performed as described previously
(21). Briefly, chromatin was
incubated overnight with p53 antibody, or no antibody added as a
negative control. The sequences of the primers were
survivin-F, 5′-TGGGTGCCCCGACGT-3′, and survivin-R,
5′-GAAGGGCCAGTTCTTGAATGTAGA-3′.
Immunofluorescence analysis
Cells were cultured in 6-well plates and then washed
with cold phosphate-buffered saline (PBS) and fixed with cold 3.7%
polyformaldehyde for 30 min. The primary antibodies were diluted
1:200 in PBS and incubated with the cells at 4°C overnight followed
by washing with 0.25% PBS-Triton X-100. Fluorescein-labeled IgG was
diluted 1:1,000 with PBS and incubated with the cells to bind with
the primary antibodies, anti-mouse IgG labeled with Cy3 (Sigma
Chemical Co., St. Louis, MO, USA) for p53, anti-rabbit IgG labeled
with FITC (Sino-American Biotechnology, Shanghai, China) for
survivin, and Hochest 33258 to stain nuclei. Cellular localization
of proteins was observed under a fluorescence microscope or by
Laser Scanning Confocal Microscopy (Leica Microsystems Inc.,
USA).
Apoptosis and cell cycle analyses by
FCM
Cultured cells were harvested and washed with 1X PBS
and then suspended in 1X PBS containing 0.1% glucose. Cold 70%
ethanol was added and mixed immediately and then the ethanol was
removed by centrifugation at 1,000 rpm. Cells were washed 2 times
with PBS, 100 μl of PC buffer were added and the solution was
incubated at room temperature for 30 min. The cells were suspended
in 100 μl PBS, 10 mg/ml RNase (10 μl), and propidium iodide
solution (10 μl) and then incubated at room temperature for 30 min.
Cells were transferred to FACS tubes and analyzed by flow
cytometry.
Statistical analysis
Data are expressed as means ± SD and statistical
comparisons were performed using the Student’s t-test. A value of
P<0.05 was considered statistically significant.
Results
LMP1 upregulated survivin expression and
its Thr34 phosphorylation by p53
We previously found that LMP1 could upregulate the
expression and activities of both p53 and survivin. We next
investigated the correlation of increased survivin and p53 mediated
by LMP1. Phosphorylated p53 (Ser20) and survivin (Thr34) were
selected to reflect p53 and survivin activity, respectively
(9,16). We found that LMP1 induced the
expression of p53 and survivin simultaneously in the LMP1-positive
CNE1 and NP69 cell lines, as well as the phosphorylation of p53
(Ser20) and survivin (Thr34) (Fig. 1A
and B). We further investigated their correlation using a
loss-of-function assay. Knockdown of p53 by siRNA showed that
survivin protein level and phosphorylated survivin (Thr34) were
decreased in p53-depleted CNE1-LMP1 and NP69-LMP1 cells (Fig. 1C and D). These data suggest that
LMP1 increases survivin expression and activity by p53 in NPC.
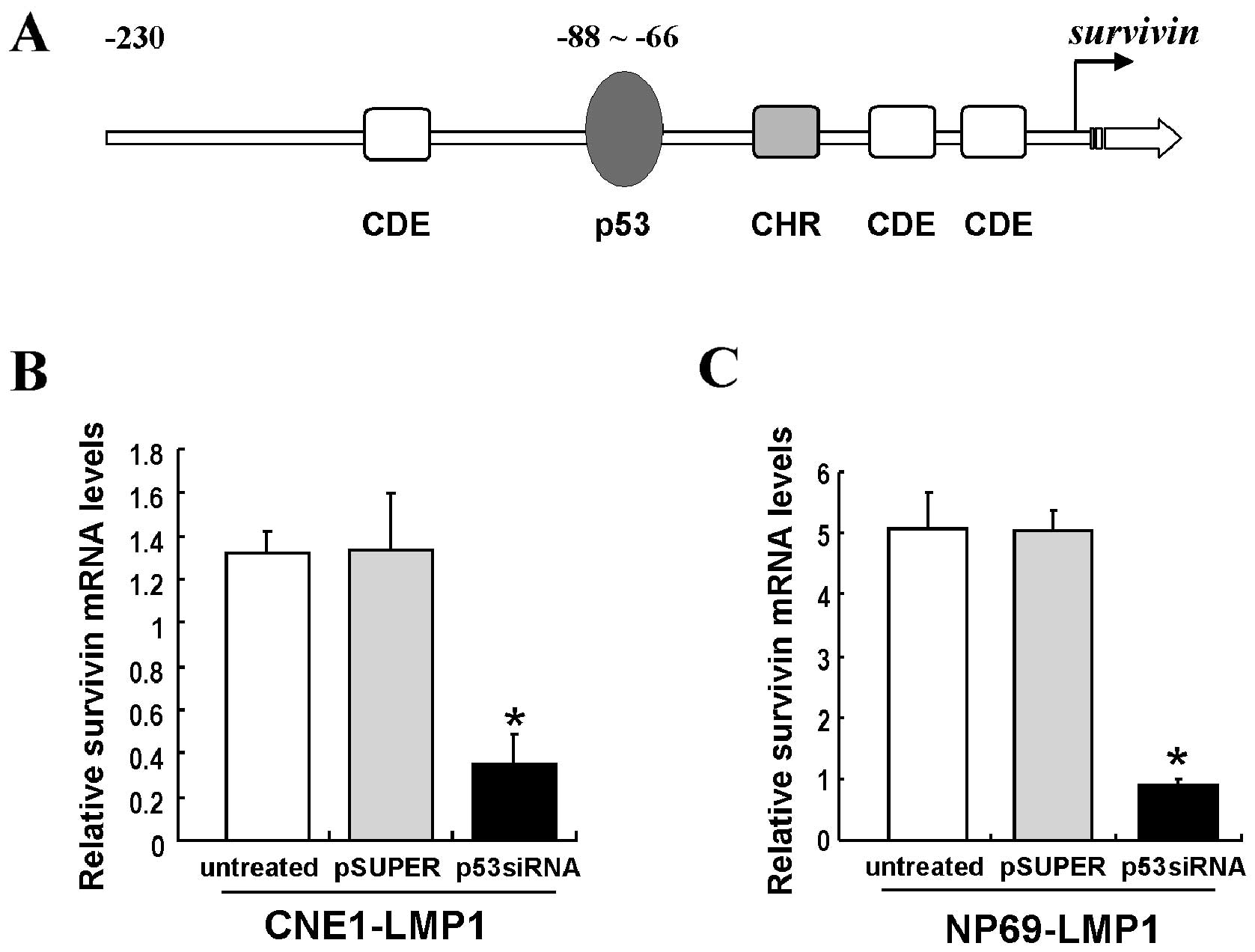
LMP1 increases survivin expression by p53
at the transcriptional level
The survivin promoter contains a p53 binding
element (Fig. 2A). We thus
further investigated whether LMP1 increased survivin expression by
p53 at the transcriptional level. Quantitative PCR (Q-PCR) showed
that survivin mRNA levels were significantly decreased in
p53-siRNA-transfected CNE1-LMP1 and NP69-LMP1 cells (P<0.05)
(Fig. 2B and C). The
survivin promoter-luciferase reporter (pGL3-Sur1.8kb) was
used to assess the effect of survivin promoter activity by
p53 in the regulation of LMP1. Results revealed that the
survivin promoter activity was significantly increased in
CNE1-LMP1 cells compared with CNE1 cells (P<0.05), while a
dramatic inhibition of the survivin promoter activity
appeared in p53-depleted CNE1-LMP1 cells but not in CNE1 cells
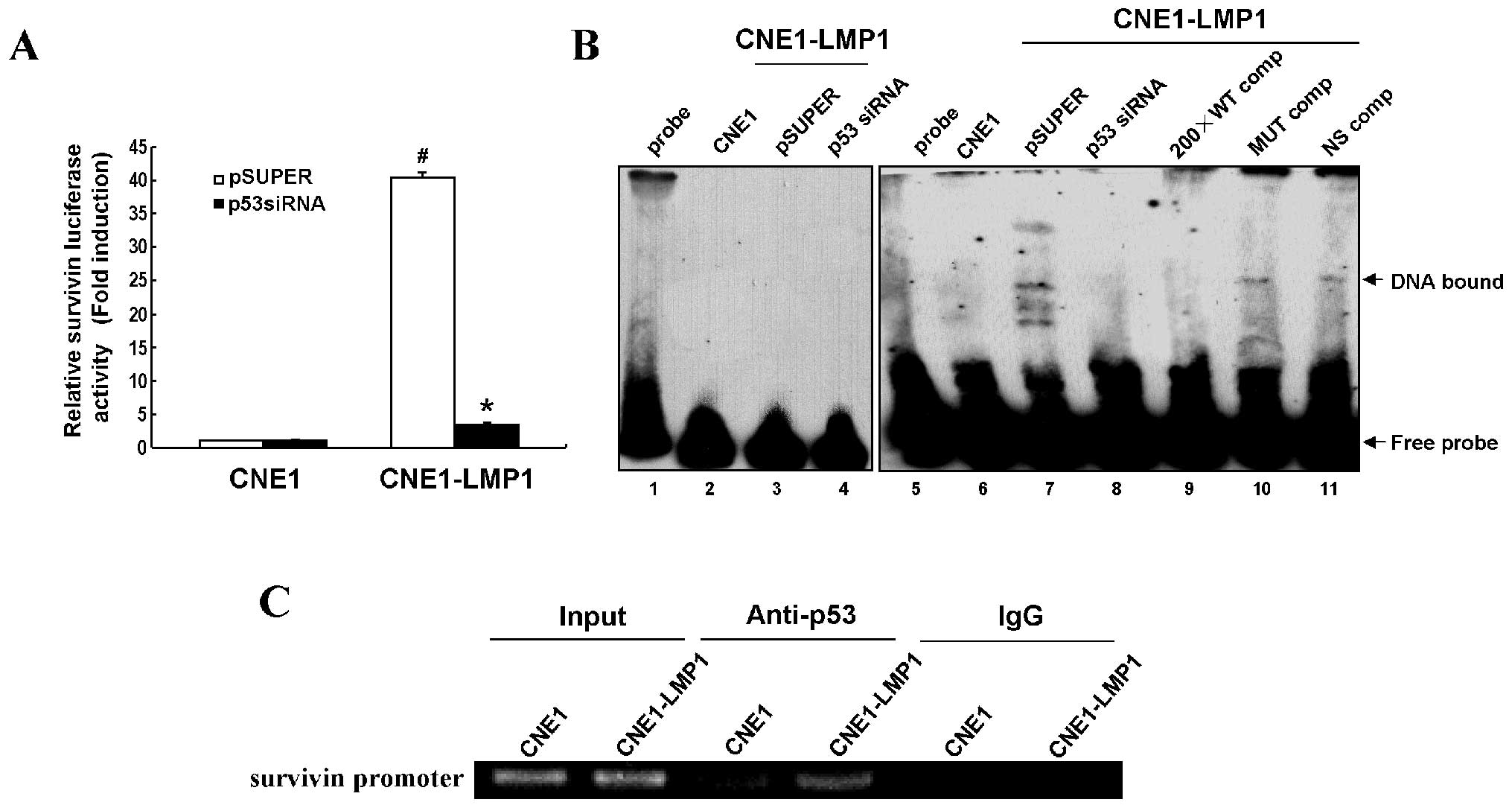
(P<0.05) (Fig. 3A).
Next, EMSA was performed to verify whether LMP1
promoted p53 binding to the survivin promoter.
Biotin-labeled wild-type or mutant p53 oligonucleotide probes were
incubated with nuclear extracts of CNE1, CNE1-LMP1 or p53-depleted
CNE1-LMP1 cells. Our data show that LMP1 obviously inhibited
p53-survivin DNA binding activity (Fig. 3B, lanes 6–8). A biotin-labeled
mutant p53 oligonucleotide probe was used as a negative control
(Fig. 3B, lanes 1–4). A 200-fold
excess of an unlabeled wild-type p53 oligonucleotide probe
efficiently inhibited the p53-DNA binding activity (Fig. 3B, lane 9), whereas an unlabeled
mutant p53 probe (Fig. 3B, lane
10) or a non-specific unlabeled p53 probe (Fig. 3B, lane 11) could not, confirming
the binding specificity of these up-shifts. ChIP assay further
demonstrated increased binding of p53 to the survivin
promoter region mediated by LMP1 in vivo (Fig. 3C). Thus, survivin may act as a
transcription target of p53 in the regulation of LMP1, responsible
for its upregulation in NPC.
LMP1 induces the co-localization of p53
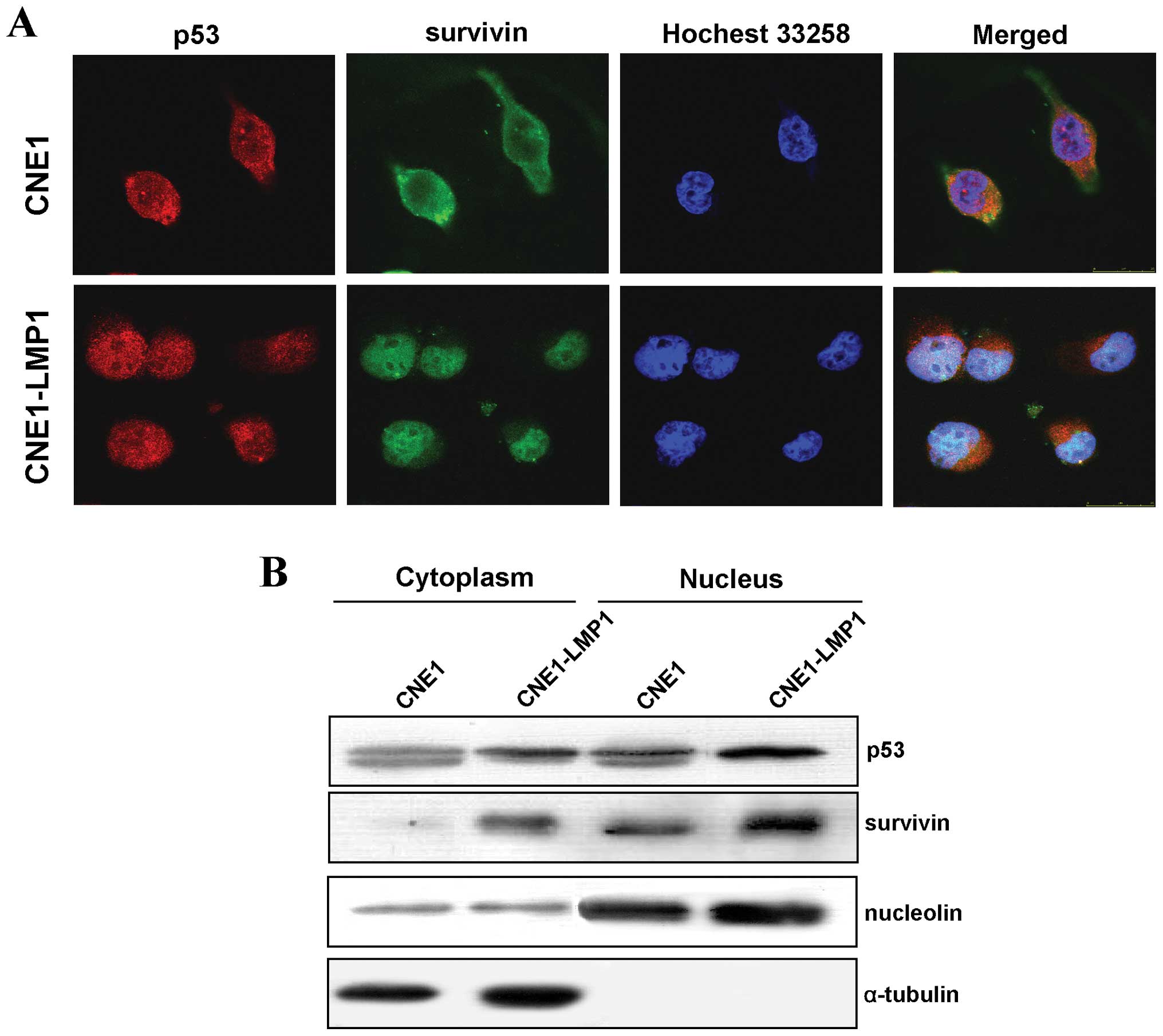
and survivin in the nucleus
Both functional p53 and survivin are located in the
nucleus. Therefore, an immunofluorescence assay was used to examine
their cellular localization. We observed that survivin and p53 were
translocated into the nucleus of CNE1-LMP1 cells cooperatively
(Fig. 4A). Western blot analyses
of cellular fractions showed that both p53 and survivin were
detected in the cytoplasm factions in CNE and CNE1-LMP1 cells, but
LMP1 obviously increased the expression of p53 and survivin in the
nucleus (Fig. 4B), confirming the
immunofluorescence results. Thus, LMP1 can promote the nuclear
accumulation of p53 and survivin, facilitating their functional
execution in NPC tumorigenesis.
Activation of p53 signaling by LMP1
results in G1/S cell cycle progression but does not induce
apoptosis
As survivin possesses a dual function in promoting
cell cycle progression and inhibiting apoptosis, and p53 is
responsible for the activation of survivin, we next investigated
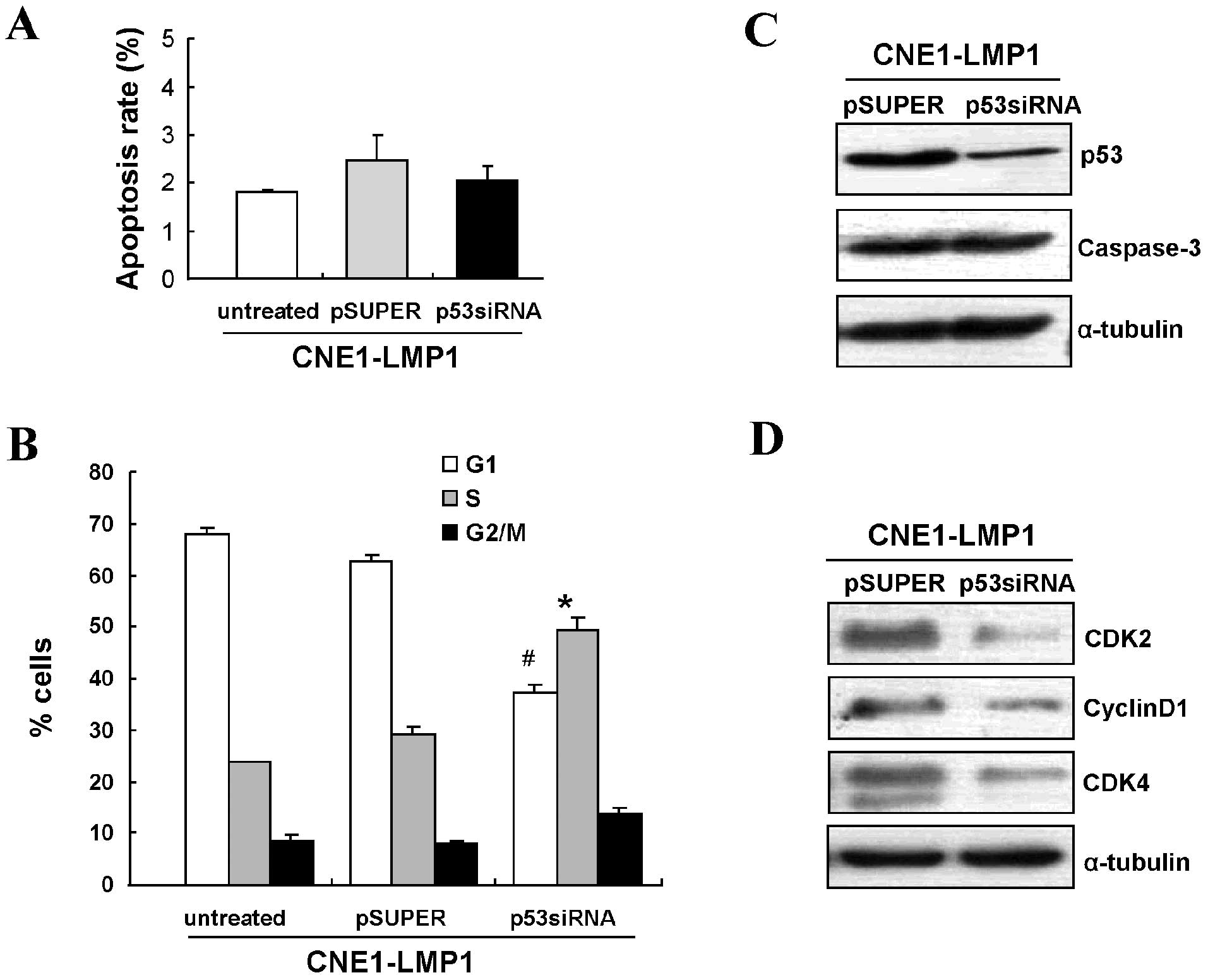
the effects of p53 on cell cycle and apoptosis mediated by LMP1
using siRNA to knockdown p53 expression. Flow cytometry showed that
knowdown of p53 minimally affected the apoptosis rate of
LMP1-positive cells (Fig. 5A).
Consistent with this result, no difference in caspase-3 protein
levels was observed in either of these two groups (Fig. 5B).
Cell cycle analyses demonstrated that targeting p53
by siRNA could increase the number of LMP1-positive cells in the S
phase (P<0.05) and decrease those in G0/G1 (P<0.05) (Fig. 5C). Western blot analysis further
confirmed that LMP1 increased the expression of G1/S checkpoint
related proteins, CDK2, CDK4 and cyclin D1 (Fig. 5D). These data suggest that
activated p53 signaling by LMP1 promotes G1/S cell cycle
progression, but not apoptosis in NPC tumorigenesis.
Discussion
p53 is well known as a tumor suppressor gene, and
its mutation has been found to be the most frequent genetic
alteration in human malignancy. However, p53 overexpression or
accumulation with a rare mutation has been identified in NPC,
unlike other types of cancer. More and more evidence demonstrates
that p53 overexpression occurs at an early stage in the development
of NPC (22) and is associated
with an advanced disease stage with a poor prognosis (23). We and others have shown that p53
can be phosphorylated and activated by LMP1, thus it might be a
transcription factor in NPC (12–15). In this study, we further studied
the potential downstream target and biological function of p53
mediated by LMP1 in NPC pathogenesis.
Survivin is a central player in regulating cell
cycle progression and apoptosis inhibition (24), and the regulation of
survivin by p53 is complicated. For examples, the p53
transcription factor directly binds the survivin promoter
alone or in combination with other protein(s), such as E2F, Sin3 or
HDAC, leading to the suppression of survivin (25,26). The p53 protein regulates survivin
phosphorylation by binding to the subunit of Cdc2/cyclin B1 kinase
(19), conversely, survivin could
regulate p53 through caspase/Mdm2 (27) and Aurora B (28). Moreover, mutated p53 can stimulate
the expression of survivin through one or more signaling pathways
(29). By knockdown of p53
protein expression with siRNA, we found that LMP1 promotes
p53-mediated survivin upregulation by increasing survivin
promoter activity and p53-survivin DNA binding activity, suggesting
the complexity of the regulation of p53 on survivin mediated by
viral oncoprotein LMP1 in NPC.
The nuclear localization is critical to the
transcriptional activity of p53 and the antiapoptotic function of
survivin. Recently, survivin was reported to be preferentially
degraded in the nucleus in the G1 phase mediated by Cdh1 (30), and forced expression of survivin
in the nucleus is sufficient to inhibit apoptosis in human cells.
Interestingly, a recent research report indicated that
overexpression of survivin in the nucleus could increase control
over the G1/S checkpoint by increasing the nuclear accumulation of
cyclin D1 and CDK4, following pRb phosphorylation, which enhanced
viral protein expression and viral replication (31). In NPC, the expression of survivin
in the nucleus is associated with poor prognosis (23). Our previous data showed that LMP1
could induce the expression of survivin and CDK4 simultaneously and
promote their co-localization in the nucleus, contributing to G1/S
cell cycle progression in NPC (32). Here, we observed that LMP1
increased nuclear localization of both p53 and survivin, required
for their function execution in NPC progression. Activated p53
signaling by LMP1 mainly promoted G1/S cell cycle progression but
did not induce apoptosis in NPC cells, consistent with our previous
findings.
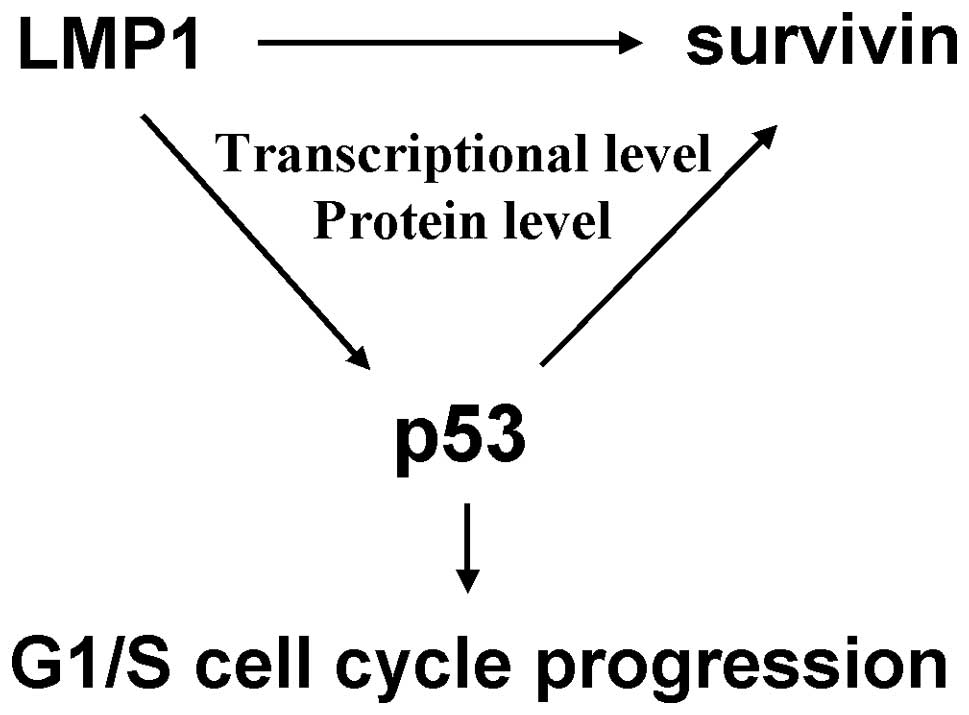
In summary, our study verified that p53 as a
transcriptional factor, could upregulate survivin expression in
both the transcriptional level and protein level mediated by LMP1,
and further stabilized the nuclear localization of survivin,
ultimately resulting in G1/S cell cycle progression but not the
induction of apoptosis in NPC (Fig.
6). These results extend our knowledge of the functional
activity and molecular mechanism of accumulated p53 in NPC
pathogenesis.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (nos. 30873010, 30801337) and the Joint
Research Fund for Overseas Chinese Scholars and Scholars in Hong
Kong and Macao (no. 81028012).
References
|
1
|
Q TaoLS YoungCB WoodmanPG
MurrayEpstein-Barr virus (EBV) and its associated human cancers -
genetics, epigenetics, pathobiology and novel therapeuticsFront
Biosci1126722713200610.2741/200016720343
|
|
2
|
Q TaoAT ChanNasopharyngeal carcinoma:
molecular pathogenesis and therapeutic developmentsExpert Rev Mol
Med9124200717477889
|
|
3
|
MA MorrisCW DawsonLS YoungRole of the
Epstein-Barr virus-encoded latent membrane protein-1, LMP1, in the
pathogenesis of nasopharyngeal carcinomaFuture
Oncol5811825200910.2217/fon.09.5319663731
|
|
4
|
SW TsaoG TramoutanisCW DawsonAK LoDP
HuangThe significance of LMP1 expression in nasopharyngeal
carcinomaSemin Cancer
Biol12473487200210.1016/S1044579X0200090112450733
|
|
5
|
H ZhengLL LiDS HuXY DengY CaoRole of
Epstein-Barr virus encoded latent membrane protein 1 in the
carcinogenesis of nasopharyngeal carcinomaCell Mol
Immunol4185196200717601372
|
|
6
|
L DengJ YangXR ZhaoCells in G2/M phase
increased in human nasopharyngeal carcinoma cell line by EBV-LMP1
through activation of NF-kappaB and AP-1Cell
Res13187194200310.1038/sj.cr.729016312862319
|
|
7
|
T FaqingH ZhiY LiqunEpstein-Barr virus
LMP1 initiates cell proliferation and apoptosis inhibition via
regulating expression of survivin in nasopharyngeal carcinomaExp
Oncol2796101200515995625
|
|
8
|
Y SunG HegamyerYJ ChengAn infrequent point
mutation of the p53 gene in human nasopharyngeal carcinomaProc Natl
Acad Sci USA8965166520199210.1073/pnas.89.14.65161631151
|
|
9
|
CH Spruck IIIYC TsaiDP HuangAbsence of p53
gene mutations in primary nasopharyngeal carcinomasCancer
Res524787479019921511442
|
|
10
|
S MuronoT YoshizakiCS ParkM
FurukawaAssociation of Epstein-Barr virus infection with p53
protein accumulation but not bcl-2 protein in nasopharyngeal
carcinomaHistopathology34432438199910.1046/j.1365-2559.1999.00625.x10231418
|
|
11
|
LF SheuA ChenHS LeeHY HsuDS YuCooperative
interactions among p53, bcl-2 and Epstein-Barr virus latent
membrane protein 1 in nasopharyngeal carcinoma cellsPathol
Int54475485200410.1111/j.1440-1827.2004.01654.x15189500
|
|
12
|
L LiS ZhouX ChenThe activation of p53
mediated by Epstein-Barr virus latent membrane protein 1 in SV40
large T-antigen transformed cellsFEBS
Lett582755762200810.1016/j.febslet.2008.01.03118242176
|
|
13
|
L LiL GuoY TaoLatent membrane protein 1 of
Epstein-Barr virus regulates p53 phosphorylation through MAP
kinasesCancer
Lett255219231200710.1016/j.canlet.2007.04.01417582679
|
|
14
|
Y SunH YiPF ZhangIdentification of
differential proteins in nasopharyngeal carcinoma cells with p53
silence by proteome analysisFEBS
Lett581131139200710.1016/j.febslet.2006.12.00817184779
|
|
15
|
Y SunH YiY YangFunctional characterization
of p53 in nasopharyngeal carcinoma by stable shRNA expressionInt J
Oncol3410171027200919287958
|
|
16
|
G AmbrosiniC AdidaDC AltieriA novel
anti-apoptosis gene, survivin, expressed in cancer and lymphomaNat
Med3917921199710.1038/nm0897-9179256286
|
|
17
|
AC MitaMM MitaST NawrockiFJ GilesSurvivin:
key regulator of mitosis and apoptosis and novel target for cancer
therapeuticsClin Cancer
Res1450005005200810.1158/1078-0432.CCR-08-074618698017
|
|
18
|
A SuzukiM HayashidaT ItoSurvivin initiates
cell cycle entry by the competitive interaction with
Cdk4/p16(INK4a) and Cdk2/cyclin E complex
activationOncogene1932253234200010.1038/sj.onc.120366510918579
|
|
19
|
DS O’ConnorD GrossmanJ PlesciaRegulation
of apoptosis at cell division by p34cdc2 phosphorylation of
survivinProc Natl Acad Sci USA971310313107200011069302
|
|
20
|
WH HoffmanS BiadeJT ZilfouJ ChenM
MurphyTranscriptional repression of the anti-apoptotic survivin
gene by wild type p53J Biol
Chem27732473257200210.1074/jbc.M10664320011714700
|
|
21
|
A MirzaM McGuirkTN HockenberryHuman
survivin is negatively regulated by wild-type p53 and participates
in p53-dependent apoptotic
pathwayOncogene2126132622200210.1038/sj.onc.120535311965534
|
|
22
|
ML GulleyMP BurtonDC AllredEpstein-Barr
virus infection is associated with p53 accumulation in
nasopharyngeal carcinomaHum
Pathol29252259199810.1016/S0046-8177(98)90044-29496828
|
|
23
|
KW YipW ShiM PintiliePrognostic
significance of the Epstein-Barr virus, p53, Bcl-2, and survivin in
nasopharyngeal cancerClin Cancer
Res1257265732200610.1158/1078-0432.CCR-06-057117020977
|
|
24
|
DC AltieriSurvivin, cancer networks and
pathway-directed drug discoveryNat Rev
Cancer86170200810.1038/nrc229318075512
|
|
25
|
K LohrC MoritzA ContenteM
Dobbelsteinp21/CDKN1A mediates negative regulation of transcription
by p53J Biol
Chem2783250732516200310.1074/jbc.M21251720012748190
|
|
26
|
M IkedaI OkamotoK TamuraDown-regulation of
survivin by ultraviolet C radiation is dependent on p53 and results
in G(2)-M arrest in A549 cellsCancer
Lett248292298200710.1016/j.canlet.2006.08.00516959403
|
|
27
|
Z WangS FukudaLM PelusSurvivin regulates
the p53 tumor suppressor gene
familyOncogene2381468153200410.1038/sj.onc.120799215361831
|
|
28
|
JE JungTK KimJS LeeSurvivin inhibits
anti-growth effect of p53 activated by aurora BBiochem Biophys Res
Commun33611641171200510.1016/j.bbrc.2005.08.23516171786
|
|
29
|
R KannangaiJ WangQZ LiuF SahinM
TorbensonSurvivin overexpression in hepatocellular carcinoma is
associated with p53 dysregulationInt J Gastrointest
Cancer355360200510.1385/IJGC:35:1:05315722574
|
|
30
|
CM ConnellR ColnaghiSP WheatleyNuclear
survivin has reduced stability and is not cytoprotectiveJ Biol
Chem28332893296200810.1074/jbc.M70446120018057009
|
|
31
|
CM ConnellSP WheatleyIA McNeishNuclear
survivin abrogates multiple cell cycle checkpoints and enhances
viral oncolysisCancer
Res6879237931200810.1158/0008-5472.CAN-08-081718829549
|
|
32
|
MD AiLL LiXR ZhaoY WuJP GongY
CaoRegulation of survivin and CDK4 by Epstein-Barr virus encoded
latent membrane protein 1 in nasopharyngeal carcinoma cell
linesCell Res15777784200510.1038/sj.cr.729034716246267
|
|
33
|
SW TsaoX WangY LiuEstablishment of two
immortalized nasopharyngeal epithelial cell lines using SV40 large
T and HPV16E6/E7 viral oncogenesBiochim Biophys
Acta1590150158200210.1016/S0167-4889(02)00208-212063178
|