Introduction
Vascular endothelial growth factor (VEGF) is a
potent mitogen displaying high specificity for vascular endothelial
cells (1). VEGF, which is
produced and secreted from a variety of cell types, increases
capillary permeability and stimulates proliferation of endothelial
cells (1). It is well known that
the metabolism of bone tissue in the mammalian skeleton is a highly
coordinated process of bone formation and bone resorption,
regulated by osteoblasts and osteoclasts, respectively (2). The microvasculature is provided by
capillary endothelial cells during bone remodeling. It is currently
recognized that the activities of osteoblasts, osteoclasts, and
capillary endothelial cells are closely coordinated, and properly
regulate bone metabolism (3).
These functional cells are considered to influence one another via
humoral factors as well as by direct cell-to-cell contact. As for
the regulation by VEGF of bone metabolism, it has been reported
that an inactivation of VEGF causes complete suppression of blood
vessel invasion concomitant with impaired trabecular bone formation
and expansion of hypertrophic chondrocyte zone in mouse tibial
epiphyseal growth plates (4).
Osteoblasts synthesize VEGF in response to various physiological
agents including transforming growth factor-β (TGF-β) (5). We have shown that the
TGF-β-stimulated VEGF synthetic cascade is positively regulated via
mitogen-activated protein (MAP) kinase superfamily such as p44/p42
MAP kinase, p38 MAP kinase and stress-activated protein
kinase/c-Jun N-terminal kinase (SAPK/JNK) in osteoblast-like
MC3T3-E1 cells (6,7). More recently, we have reported that
Rho kinase negatively regulates the TGF-β-stimulated VEGF synthesis
in MC3T3-E1 cells (8). These
findings lead us to speculate that various kinds of intracellular
molecules could modulate TGF-β-stimulated VEGF synthesis in
osteoblasts. However, the exact mechanism underlying VEGF synthesis
in osteoblasts has not yet been clarified.
Adenosine 5′-monophosphate-activated protein kinase
(AMPK) is generally known to regulate multiple metabolic pathways
(9). AMPK, defined as a mammalian
protein kinase that was allosterically activated by AMP and was
able to phosphorylate and inactivate enzymes of lipid synthesis
(10), has emerged over the last
decade as a key sensing mechanism in the regulation of cellular
energy homeostasis (11–13). AMPK is activated in mammalian
cells by a variety of physiological and pathological stresses that
increase the intracellular AMP: ATP ratio, either by increasing ATP
consumption or by decreasing ATP production. Compound C, a
pyrrazolopyrimidine derivative which competitively inhibits AMPK,
has been widely used as a specific and reversible AMPK inhibitor
(14–16).
Activated AMPK acts to restore cellular energy
balance by ATP generating pathways such as fatty acid oxidation,
while simultaneously inhibiting ATP utilizing pathways. In addition
to these functions as a metabolic regulator, it has been
demonstrated that activated AMPK regulates bone formation and bone
mass in vitro (17).
However, the precise function of AMPK in the regulation of bone
metabolism has not been fully elucidated.
In the present study, we investigated whether AMPK
is involved in the TGF-β-stimulated VEGF synthesis in osteoblasts.
We here show that AMPK is involved in TGF-β-stimulated VEGF
synthesis in osteoblasts, and that AMPK acts at a point upstream of
MEK1/2.
Materials and methods
Materials
Normal human osteoblasts (NHOst) were purchased from
Cambrex (Charles, IA). TGF-β and mouse or human VEGF enzyme-linked
immunosorbent assay (ELISA) kit were purchased from R&D
Systems, Inc. (Minneapolis, MN). Compound C, a pyrrazolopyrimidine
derivative which competitively inhibits AMPK (16), was purchased from Calbiochem, Inc.
(San Diego, CA). Phospho-specific AMPKα (Thr172) and (Ser485)
antibodies, AMPKα antibodies, phospho-specific AMPKβ (Ser108) and
(Ser182) antibodies, AMPKβ, phospho-specific Smad2, Smad2,
phospho-specific p44/p42 MAP kinase, p44/p42 MAP kinase,
phospho-specific MEK1/2 and MEK1/2 antibodies were purchased from
Cell Signaling Technology, Inc. (Beverly, MA).
Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibodies were
obtained form Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). The
ECL western blotting detection system was purchased from GE
Healthcare UK Ltd. (Buckinghamshire, UK). Other materials and
chemicals were obtained from commercial sources.
Cell culture
Cloned osteoblast-like MC3T3-E1 cells derived from
newborn mouse calvaria (18) were
maintained as previously described (19). Briefly, the cells were cultured in
α-minimum essential medium (α-MEM) containing 10% fetal calf serum
(FCS) at 37°C in a humidified atmosphere of 5% CO2/95%
air. The cells were seeded into 35-mm diameter dishes
(5×104/dish) or 90-mm diameter dishes
(20×104/dish) in α-MEM containing 10% FCS. After 5 days,
the medium was exchanged to α-MEM containing 0.3% FCS and incubated
for 48 h. The cells were then used for subsequent experiments.
NHOst cells were seeded into 35-mm
(5×104/dish) diameter dishes in α-MEM containing 10% FCS
(20). After 6 days, the medium
was changed to α-MEM containing 0.3% FCS. The cells were used for
experiments after 48 h.
VEGF assay
The cultured cells were pretreated with various
doses of compound C for 60 min, and then stimulated by 5 ng/ml
TGF-β or vehicle in 1 ml of α-MEM containing 0.3% FCS for 48 h. The
conditioned medium was collected at the end of the incubation, and
the VEGF concentration was measured by cell species-responsible
ELISA kits. The absorbance of enzyme-linked immunosorbent assay
(ELISA) samples was measured at 450 nm with the EL 340 Bio Kinetic
Reader (Bio-Tek Instruments, Inc., Winooski, VT) according to the
manufacturer’s protocol.
Real-time RT-PCR
The cultured cells were pretreated with 10 μM
compound C or vehicle for 60 min, and then stimulated by 5 ng/ml
TGF-β in a-MEM containing 0.3% FCS for the indicated periods.
Total-RNA was isolated and transcribed into complementary DNA using
TRIzol reagent (Invitrogen Corp., Carlsbad, CA) and Omniscript
reverse transcriptase kit (Qiagen, Inc., Valencia, CA),
respectively. Real-time RT-PCR was performed using a Light Cycler
system in capillaries and FastStart DNA Master SYBR-Green I
provided with the kit (Roche Diagnostics, Basel, Switzerland).
Sense and antisense primers were synthesized based on the report of
Simpson et al (21) for
mouse VEGF mRNA. Sense and antisense primers for mouse VEGF mRNA
were purchased from Takara Bio., Inc. (Tokyo, Japan) (primer set
ID: MA039013). The amplified products were determined by melting
curve analysis and agarose electrophoresis. VEGF mRNA levels were
normalized with those of GAPDH mRNA.
Western blot analysis
Western blot analysis was performed as previously
described (22). In brief, the
cultured cells were pretreated with various doses of compound C for
60 min, and then stimulated by 5 ng/ml TGF-β or vehicle in α-MEM
containing 0.3% FCS for the indicated periods. The cells were
washed twice with phosphate-buffered saline and then lysed,
homogenized and sonicated in a lysis buffer containing 62.5 mM
Tris/HCl, pH 6.8, 3% sodium dodecyl sulfate (SDS), 50 mM
dithiothreitol and 10% glycerol. SDS-polyacrylamide gel
electrophoresis (PAGE) was performed according to Laemmli (23) in 10% polyacrylamide gels. The
protein (10 μg) was fractionated and transferred onto an
Immuno-Blot PVDF membrane (Bio-Rad, Hercules, CA). Membranes were
blocked with 5% fat-free dry milk in Tris-buffered saline-Tween-20
(TBS-T; 20 mM Tris/HCl, pH 7.6, 137 mM NaCl, 0.1% Tween-20) for 2 h
before incubation with the primary antibodies. Peroxidase-labeled
antibodies raised in goat against rabbit IgG were used as second
antibodies. The primary and secondary antibodies were diluted at
1:1,000 with 5% fat-free dry milk in TBS-T. Peroxidase activity on
the membrane was visualized on X-ray film by means of the ECL
western blotting detection system.
Statistical analysis
The data were analyzed by ANOVA followed by the
Bonferroni method for multiple comparisons between pairs, and a
P<0.05 was considered statistically significant. All data are
presented as the mean ± SEM of triplicate independent
determinations.
Results
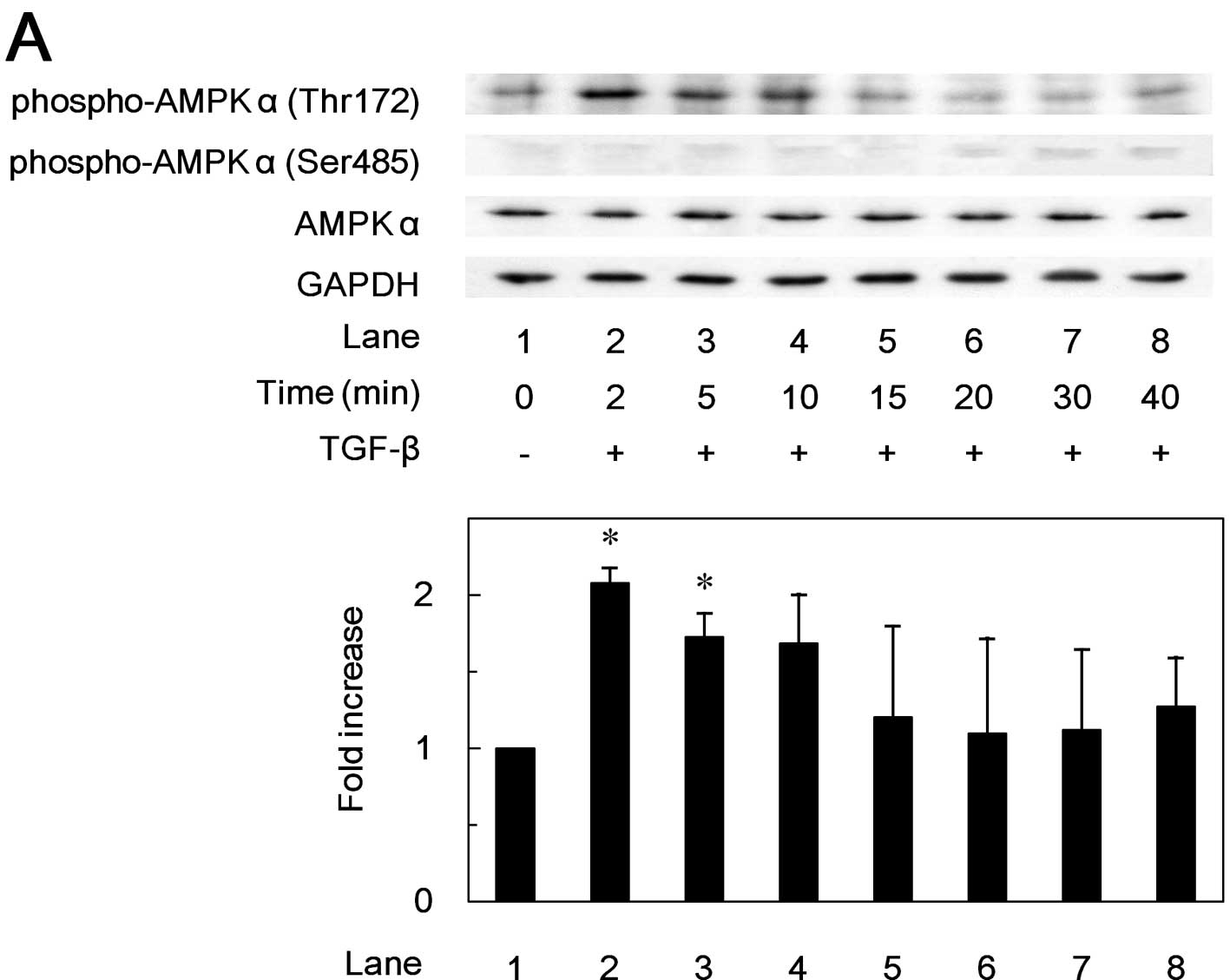
Effects of TGF-β on the phosphorylation
of AMPK subunits in MC3T3-E1 cells
In order to investigate whether TGF-β stimulates the
activation of AMPK in osteoblast-like MC3T3-E1 cells, we first
examined the effects of TGF-β on the phosphorylation of the AMPK
subunits. TGF-β markedly induced the phosphorylation of AMPK
α-subunit (Thr172) in a time-dependent manner (Fig. 1A). The phosphorylation of the AMPK
α-subunit (Thr172) by TGF-β reached its maximum at 2 min, and
decreased thereafter. TGF-β also time-dependently induced the
phosphorylation of the AMPK β-subunit (Ser108) (Fig. 1B). The phosphorylation level of
AMPK β-subunit (Ser108) reached a peak at 15 min, and decreased
thereafter. However, TGF-β had little effect on the phosphorylation
of AMPK α-subunit (Ser485) or AMPK β-subunit (Ser182) (Fig. 1).
Effect of compound C on the
TGF-β-stimulated VEGF synthesis in MC3T3-E1 cells
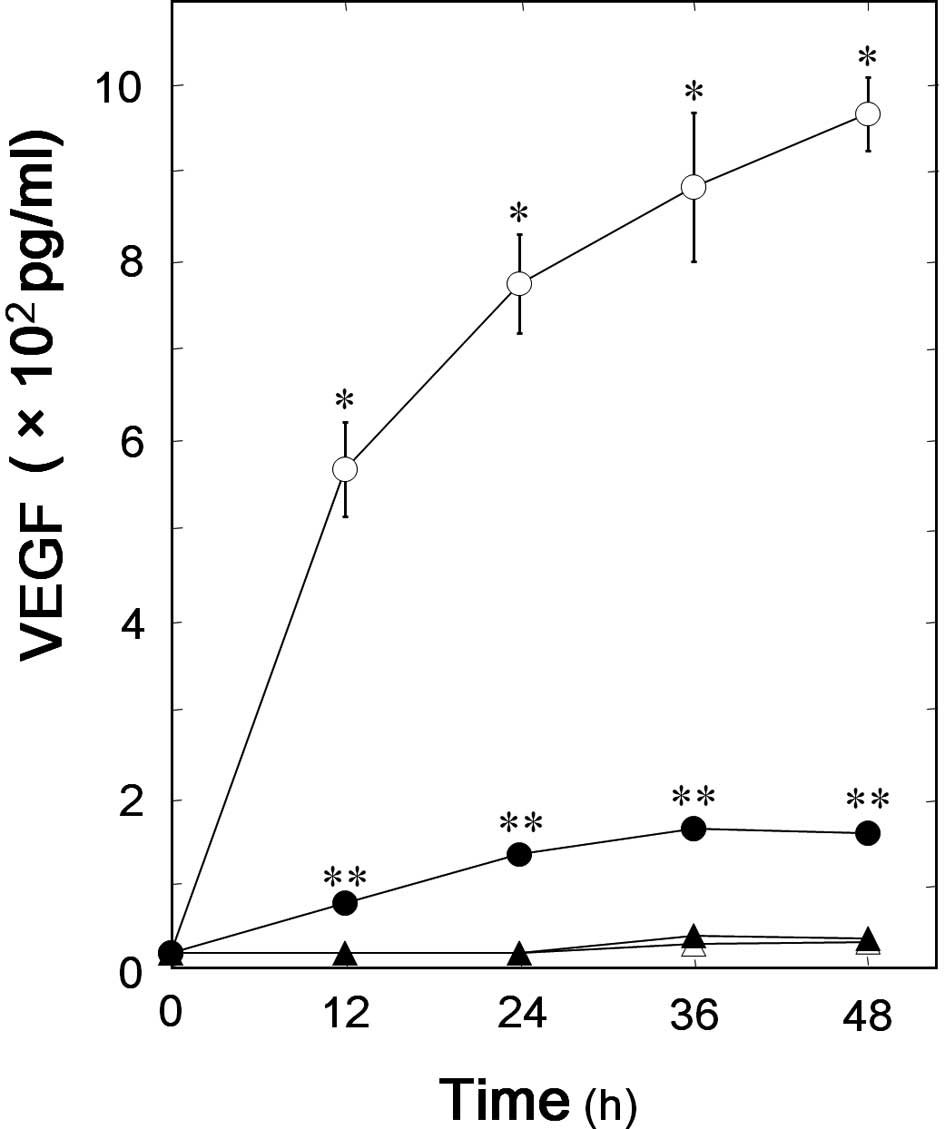
We have previously showed that TGF-β significantly
stimulates VEGF synthesis in osteoblast-like MC3T3-E1 cells
(6). To investigate whether AMPK
is involved in the TGF-β-stimulated VEGF synthesis in MC3T3-E1
cells, we next examined the effect of compound C, an inhibitor of
AMPK, on VEGF synthesis stimulated by TGF-β. Compound C
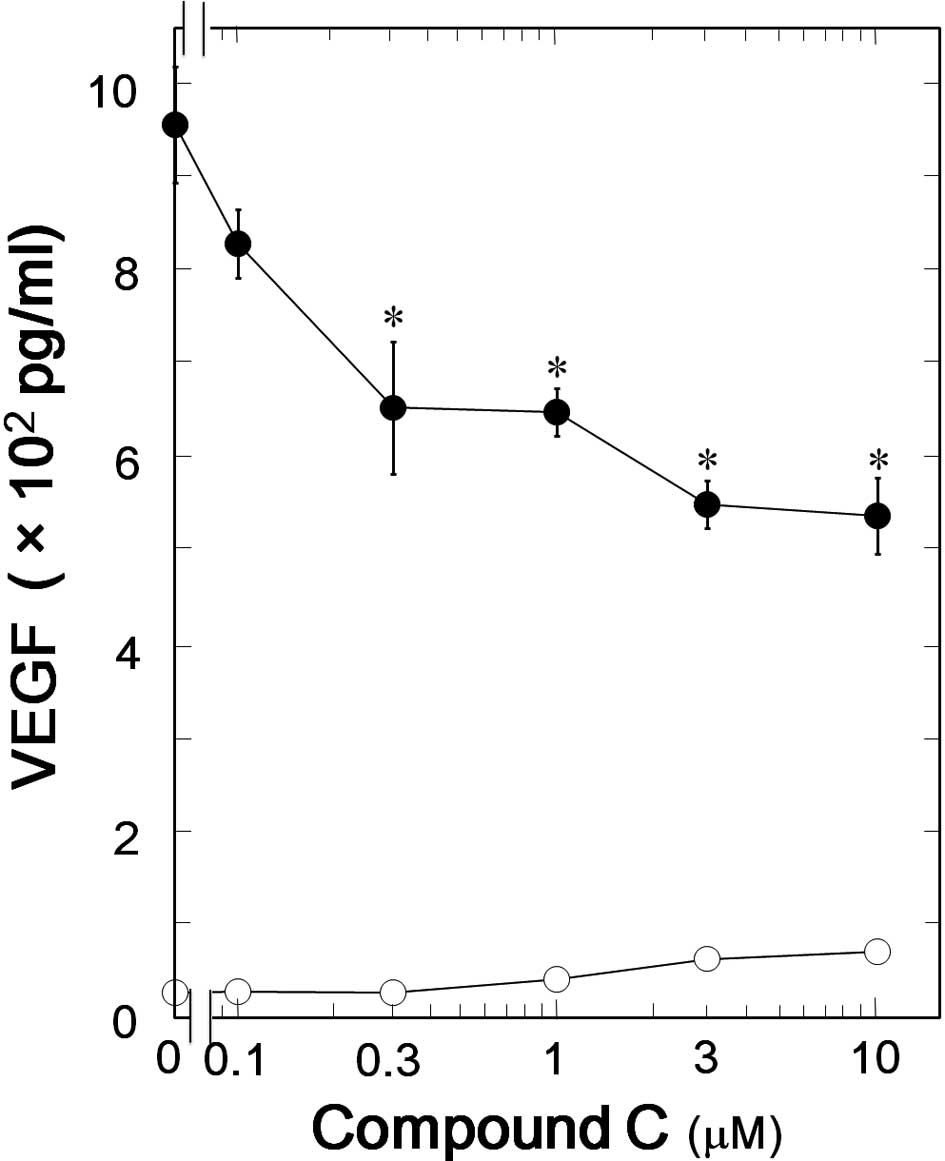
significantly suppressed the TGF-β-induced VEGF synthesis (Fig. 2). The inhibitory effect of
compound C on the VEGF synthesis was dose-dependent in the range
between 1 and 10 μM (Fig. 3). The
maximum effect of compound C was observed at 10 μM and caused
approximately 40% suppression in the TGF-β-effect.
Effect of compound C on the
TGF-β-stimulated VEGF synthesis in NHOst cells
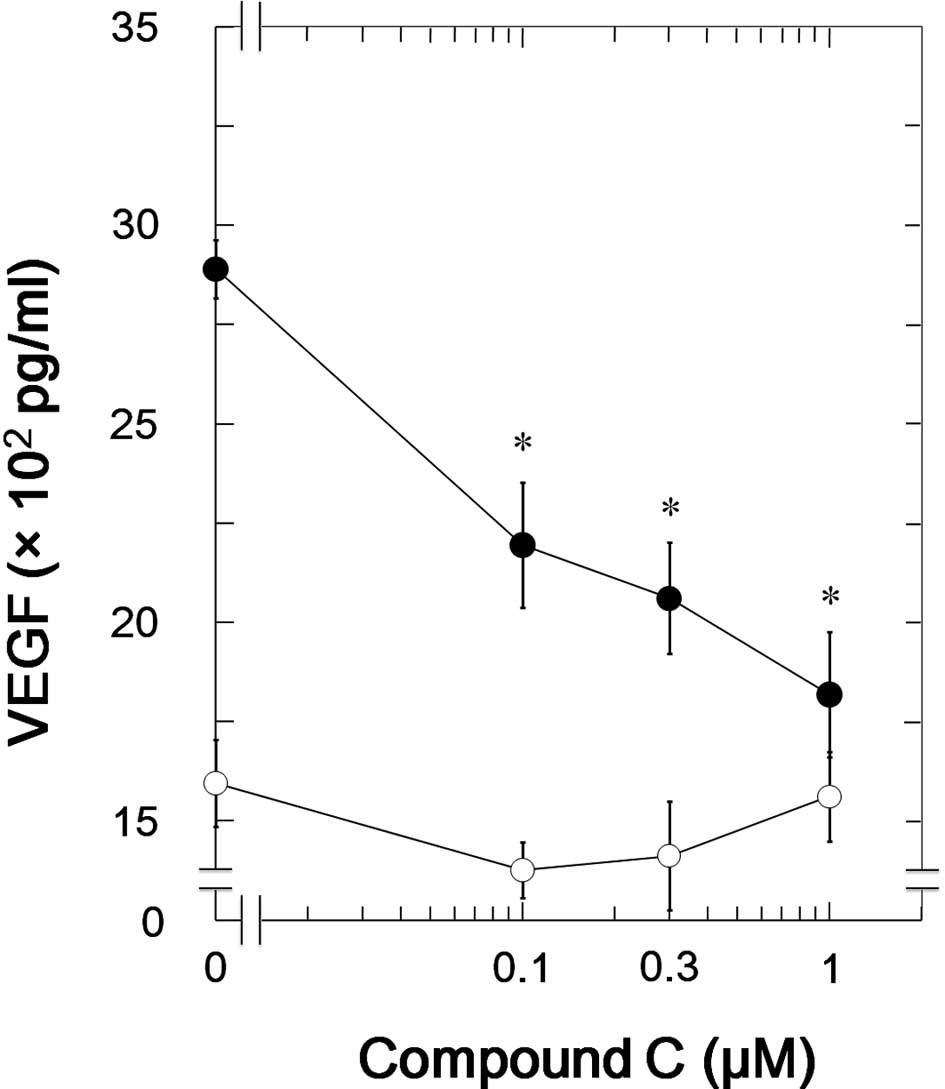
We next examined the effects of compound C in NHOst,
a different osteoblastic cell line. Compound C, as well as in
MC3T3-E1 cells, significantly suppressed the TGF-β-induced VEGF
synthesis in NHOst (Fig. 4). The
inhibitory effect of compound C on the VEGF synthesis was
dose-dependent in the range between 0.1 and 1 μM. The maximum
effect of compound C was observed at 1 μM and caused approximately
80% suppression in the TGF-β-effect.
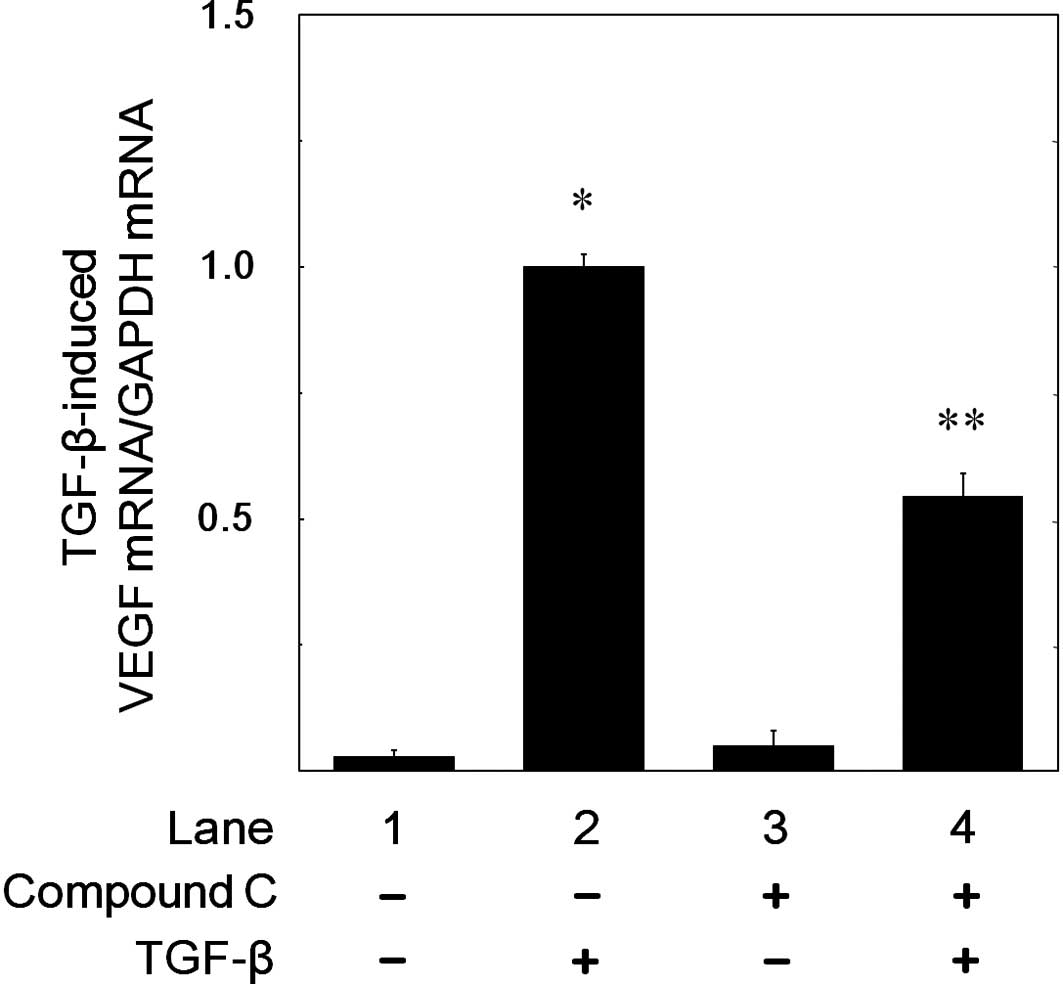
Effect of compound C on the TGF-β-induced
expression levels of VEGF mRNA in MC3T3-E1 cells
It has previously been reported that TGF-β induces
VEGF mRNA expression in MC3T3-E1 cells (5). In order to clarify whether AMPK
affects TGF-β-stimulated VEGF release through the modulation of a
transcriptional event or not, we furthermore examined the effect of
compound C on the TGF-β-induced VEGF mRNA expression. We confirmed
that VEGF mRNA expression levels induced by TGF-β were increased in
a time-dependent manner in accordance with a previous report
(5). Compound C (10 μM)
significantly suppressed the TGF-β-induced VEGF mRNA expression
levels (Fig. 5). Compound C of 10
μM caused approximately 50% inhibition in the TGF-β-effect. We
confirmed that GAPDH mRNA was constitutively expressed and stable
in MC3T3-E1 cells (data not shown).
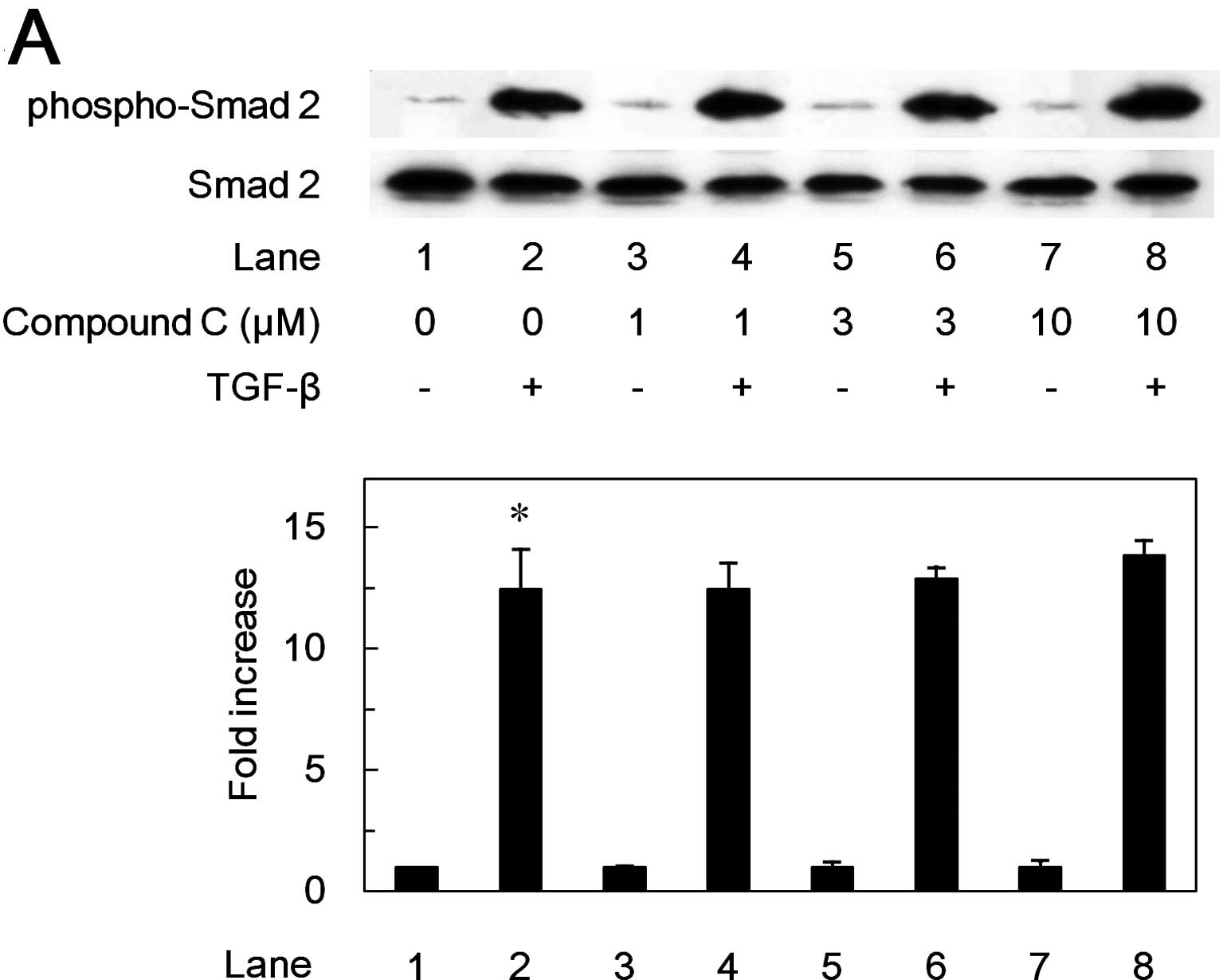
Effects of compound C on the
phosphorylation of Smad2 or p44/p42 MAP kinase induced by TGF-β in
MC3T3-E1 cells
It is well known that receptor-activated Smads such
as Smad2 are principal mediators of intracellular signaling from
TGF-β receptors to the nucleus (24). Thus, we next examined the effect
of compound C on the TGF-β-induced phosphorylation of Smad2.
However, compound C failed to affect the TGF-β-induced
phosphorylation level of Smad2 in the range between 1 and 10 μM
(Fig. 6A).
It is currently recognized that other proteins
mediate the intracellular signaling by TGF-β in addition to Smads
(25). We have previously
demonstrated that p44/p42 MAP kinase, p38 MAP kinase and SAPK/JNK
are involved in the TGF-β-stimulated VEGF synthesis in
osteoblast-like MC3T3-E1 cells (6,7).
In addition, we examined the effects of compound C on the
TGF-β-induced phosphorylation of p44/p42 MAP kinase, p38 MAP kinase
or SAPK/JNK. Compound C markedly suppressed the TGF-β-induced
phosphorylation level of p44/p42 MAP kinase in a dose-dependent
manner between 1 and 10 μM (Fig.
6B). Furthermore, compound C also suppressed TGF-β-stimulated
phosphorylation of MEK1/2, an upstream kinase that activates
p44/p42 MAPK (Fig. 6C). On the
other hand, compound C had little effect on the phosphorylation of
p38 MAP kinase or SAPK/JNK induced by TGF-β (data not shown).
Discussion
AMPK exists as heterotrimeric complex comprising a
catalytic α-subunit and regulatory β- and γ-subunits (9). Phosphorylation of the Thr172 residue
in the α-subunit is essential for AMPK activation (26). Once activated, AMPK causes an over
100-fold increase in kinase activity (27,28). A carbohydrate binding module (CBM)
is located within the central region of the β-subunit (9). In the CBM, multiple
autophosphorylation sites including Ser108 have been identified
(29). In the present study, we
demonstrated that the maximum phosphorylation of the AMPK α-subunit
(Thr172) occurred at 5 min, followed by a peak phosphorylation of
the AMPK β-subunit (Ser108) at 15 min. Although further examination
is warranted to elucidate the precise mechanism of the relationship
between AMPK activation and TGF-β stimulation, our results strongly
suggest that TGF-β triggers phosphorylation of AMPK followed by a
series of activation events of AMPK for TGF-β-stimulated VEGF
synthesis in osteoblasts. In addition, we found that compound C
(15,16) also suppressed TGF-β-induced VEGF
synthesis in NHOst cells. It is likely that AMPK is involved in
TGF-β-induced VEGF synthesis also on human osteoblasts. To the best
of our knowledge, this is the first report showing the involvement
of AMPK in TGF-β-stimulated VEGF synthesis in osteoblasts.
It is well known that TGF-β mainly employs
receptor-activated Smad proteins such as Smad2 and Smad3 as
intracellular mediators of signaling (30). However, compound C, an AMPK
inhibitor, had no effect on the TGF-β-induced phosphorylation of
Smad2 in osteoblast-like MC3T3-E1 cells. Therefore, it seems
unlikely that AMPK acts at a point upstream of Smad2 in the VEGF
synthesis in MC3T3-E1 cells. It is currently recognized that TGF-β
exerts its effects on a variety of biological functions via
Smad-independent signaling in addition to Smad-dependent signaling
(25). The MAP kinase
superfamily, such as the p44/p42 MAP kinase, p38 MAP kinase and
SAPK/JNK act as central elements used by mammalian cells to
transduce the various extracellular messages (24,31). As for TGF-β-stimulated VEGF
synthesis in osteoblast-like MC3T3-E1 cells, we have reported the
involvement of p44/p42 MAP kinase, p38 MAP kinase and SAPK/JNK
(6,7). Thus, we investigated the effects of
compound C on the TGF-β-induced phosphorylation of these three MAP
kinases. Compound C had little effect on the phosphorylation of
SAPK/JNK or p38 MAP kinase induced by TGF-β. In contrast,
TGF-β-induced phosphorylation of both p44/42 MAP kinase and MEK1/2
was markedly suppressed by compound C. These findings strongly
suggest that AMPK acts at a point upstream of MEK1/2 in the
TGF-β-stimulated VEGF synthesis in osteoblast-like MC3T3-E1 cells.
TGF-β-activated kinase (TAK1), a member of the MAP kinase kinase
kinase family, has been identified as an upstream kinase of MAP
kinase including osteoblast-like MC3T3-E1 cells (32). It has been recently reported that
expression of TAK1 in HeLa cells stimulates phosphorylation of the
AMPK α-subunit (Thr172) (33).
Based on these findings, it is most likely that AMPK may act as a
regulator at the point between TAK1 and MEK1/2 in TGF-β-stimulated
VEGF synthesis in osteoblast-like MC3T3-E1 cells. Recent reports
have indicated that the possibility of AMPK-independent effects
exerted by compound C (16,34,35). Thus, it is possible that an
AMPK-independent mechanism is involved in compound C-induced
suppression of TGF-β-stimulated VEGF synthesis in osteoblasts.
However, there is no available inhibitor for AMPK which is more
specific than compound C to the best of our knowledge. Further
investigations would be required to clarify the details.
Recent studies have reported that both AMPK and VEGF
play a significant role in bone metabolism (1,17,36,37). Germline deletion of either AMPKβ1
or β2 isoforms reportedly resulted in reduced trabecular bone
density and mass without effects on osteoclast or osteoblast
numbers, showing that AMPK is required to maintain normal bone
density (38). Therefore, it is
possible that TGF-β-induced VEGF synthesis via MEK1/2-p44/p42 MAP
kinase regulated by AMPK plays an important role in the homeostasis
of bone density under physiological conditions. Many hormones and
neuromediators, including leptin, ghrelin, cannabinoids, and the
sympathetic nervous system that regulate food intake and energy
expenditure through AMPK activation also regulate bone mass
(39–47). VEGF is critical for bone
angiogenesis, and VEGF secreted from osteoblasts may play a pivotal
role in the regulation of bone metabolism. It has been reported
that TGF-β induces differentiation or proliferation of osteoblasts,
and inhibits the formation of osteoclast precursors (48). Therefore, it is highly speculated
that TGF-β-stimulated VEGF synthesis via AMPK acts as a positive
regulator of bone remodeling and AMPK is probably a key molecule in
bone metabolism as seen in fat metabolism. We have recently
reported that AMPK positively regulates FGF-2-stimulated VEGF
synthesis via both p44/p42 MAP kinase and SAPK/JNK in
osteoblast-like MC3T3-E1 cells (49). It is likely that the regulatory
mechanisms of VEGF synthesis by AMPK in osteoblasts are different
in each stimulus, suggesting that the sophisticated regulation by
AMPK is essential to promote the cellular event, VEGF synthesis.
However, the exact role of AMPK in osteoblasts is not fully
clarified. Further investigation is necessary to elucidate the role
of AMPK in bone metabolism.
In conclusion, our results strongly suggest that
AMPK functions at a point upstream of MEK1/2 and positively
regulates TGF-β-stimulated VEGF synthesis via p44/p42 MAP kinase
activation in osteoblasts.
Acknowledgements
We are very grateful to Yoko Kawamura for her
skillful technical assistance. This investigation was supported in
part by a Grant-in-Aid for Scientific Research (19591042) from the
Ministry of Education, Science, Sports and Culture of Japan, the
Foundation for Growth Science, and by Research Grants for Longevity
Sciences (21A-4, 21A-22) from the Ministry of Health, Labour and
Welfare of Japan.
References
|
1
|
N FerraraVascular endothelial growth
factor: basic science and clinical progressEndocr
Rev25581611200410.1210/er.2003-002715294883
|
|
2
|
G KarsentyEF WagnerReaching a genetic and
molecular understanding of skeletal developmentDev
Cell2389406200210.1016/S1534-5807(02)00157-011970890
|
|
3
|
A ErlebacherEH FilvaroffSE GitelmanR
DerynckToward a molecular understanding of skeletal
developmentCell80371378199510.1016/0092-8674(95)90487-57859279
|
|
4
|
E ZelzerBR OlsenMultiple roles of vascular
endothelial growth factor (VEGF) in skeletal development, growth,
and repairCurr Top Dev
Biol65169187200510.1016/S0070-2153(04)65006-X15642383
|
|
5
|
PB SaadehBJ MehraraDS SteinbrechME
DudziakJA GreenwaldJS LuchsJA SpectorH UenoGK GittesMT
LongakerTransforming growth factor-β1 modulates the expression of
vascular endothelial growth factor by osteoblastsAm J
Physiol277C628C6371999
|
|
6
|
H TokudaD HatakeyamaS AkamatsuK TanabeM
YoshidaT ShibataO KozawaInvolvement of MAP kinases in
TGF-β-stimulated vascular endothelial growth factor synthesis in
osteoblastsArch Biochem Biophys4151171252003
|
|
7
|
Y KannoA IshisakiM YoshidaH TokudaO
NumataO KozawaSAPK/JNK plays a role in transforming growth
factor-β-induced VEGF synthesis in osteoblastsHorm Metab
Res371401452005
|
|
8
|
M KunoS TakaiR Matsushima-NishiwakiC
MinamitaniJ MizutaniT OtsukaA HaradaS AdachiO KozawaH
TokudaRho-kinase inhibitors decrease TGF-β-stimulated VEGF
synthesis through stress-activated protein kinase/c-Jun N-terminal
kinase in osteoblastsBiochem Pharmacol771962032009
|
|
9
|
S FogartyDG HardieDevelopment of protein
kinase activators: AMPK as a target in metabolic disorders and
cancerBiochim Biophys
Acta1804581591201010.1016/j.bbapap.2009.09.01219778642
|
|
10
|
LA YehKH LeeKH KimRegulation of rat liver
acetyl-CoA carboxylase. Regulation of phosphorylation and
inactivation of acetyl-CoA carboxylase by the adenylate energy
chargeJ Biol Chem2552308231419806102090
|
|
11
|
DG HardieSA HawleyJW ScottAMP-activated
protein kinase - development of the energy sensor conceptJ
Physiol574715200610.1113/jphysiol.2006.10894416644800
|
|
12
|
R LageC DieguezA Vidal-PuigM LopezAMPK. a
metabolic gauge regulating whole-body energy homeostasisTrends Mol
Med14539549200810.1016/j.molmed.2008.09.00718977694
|
|
13
|
GR SteinbergBE KempAMPK in health and
diseasePhysiol
Rev8910251078200910.1152/physrev.00011.200819584320
|
|
14
|
G ZhouR MyersY LiY ChenX ShenJ
Fenyk-MelodyM WuJ VentreT DoebberN FujiiRole of AMP-activated
protein kinase in mechanism of metformin activaitonJ Clin
Invest10811671174200110.1172/JCI1350511602624
|
|
15
|
EK KimI MillerS AjaJE LandreeM PinnC75, a
fatty acid synthase inhibitor, reduces food intake via hypothalamic
AMP-activated protein kinaseJ Biol
Chem2791997019976200410.1074/jbc.M40216520015028725
|
|
16
|
M NamWH LeeEJ BaeSG KimCompound C inhibits
clonal expansion of preadipocytes by increasing p21 level
irrespectively of AMPK inhibitionArch Biochem
Biophys4797481200810.1016/j.abb.2008.07.02918721791
|
|
17
|
M ShahB KolaA BataveljicTR ArnettB
ViolletL SaxonM KorbonitsC ChenuAMP-activated protein kinase (AMPK)
activation regulates in vitro bone formation and bone
massBone47309319201010.1016/j.bone.2010.04.59620399918
|
|
18
|
H SudoHA KodamaY AmagaiS YamamotoS KasaiIn
vitro differentiation and calcification in a new clonal osteogenic
cell line derived from newborn mouse calvariaJ Cell
Biol96191198198310.1083/jcb.96.1.1916826647
|
|
19
|
O KozawaH TokudaM MiwaJ KotoyoriY
OisoCross-talk regulation between cyclic AMP production and
phosphoinositide hydrolysis induced by prostaglandin E2 in
osteoblast-like cellsExp Cell
Res198130134199210.1016/0014-4827(92)90158-51309194
|
|
20
|
H NatsumeH TokudaS AdachiS TakaiR
Matsushima-NishiwakiK KatoC MinamitaniS NiidaJ MizutaniO
KozawaRho-kinase limits FGF-2-stimulated VEGF release in
osteoblastsBone4610681074201010.1016/j.bone.2010.01.37820114091
|
|
21
|
DA SimpsonS FeeneyC BoyleAW StittRetinal
VEGF mRNA measured by SYBR green I fluorescence: a versatile
approach to quantitative PCRMol Vis6178183200011023552
|
|
22
|
K KatoH ItoK HasegawaY InagumaO KozawaT
AsanoModulation of the stress-induced synthesis of hsp27 and
αB-crystallin by cyclic AMP in C6 rat glioma cellsJ
Neurochem669469501996
|
|
23
|
UK LaemmliCleavage of structural proteins
during the assembly of the head of bacteriophage
T4Nature227680685197010.1038/227680a05432063
|
|
24
|
JM KyriakisJ AvruchMammalian
mitogen-activated protein kinase signal transduction pathways
activated by stress and inflammationPhysiol
Rev81807889200111274345
|
|
25
|
A MoustakasCH HeldinNon-Smad TGF-β
signalsJ Cell Sci118357335842005
|
|
26
|
SA HawleyM DavisonA WoodsSP DaviesRK BeriD
CarlingDG HardieCharacterization of the AMP-activated protein
kinase kinase from rat liver and identification of threonine 172 as
the major site at which it phosphorylates AMP-activated protein
kinaseJ Biol
Chem2712787927887199610.1074/jbc.271.44.278798910387
|
|
27
|
SC SteinA WoodsNA JonesMD DavisonD
CarlingThe regulation of AMP-activated protein kinase by
phosphorylationBiochem
J345437443200010.1042/0264-6021:345043710642499
|
|
28
|
M SuterU RiekR TuerkU SchlattnerT
WallimannD NeumannDissecting the role of 5′-AMP for allosteric
stimulation, activation, and deactivation of AMP-activated protein
kinaseJ Biol Chem28132207322162006
|
|
29
|
KI MitchelhillBJ MichellCM HouseD
StapletonJ DyckJ GambleC UllrichLA WittersBE KempPosttranslational
modifications of the 5′-AMP-activated protein kinase β1 subunitJ
Biol Chem27224475244791997
|
|
30
|
K MiyazawaM ShinozakiT HaraT FuruyaK
MiyazonoTwo major Smad pathways in TGF-β superfamily
signallingGenes Cells7119111042002
|
|
31
|
C WidmannS GibsonMB JarpeGL
JohnsonMitogen-activated protein kinase: conservation of a
three-kinase module from yeast to humanPhysiol
Rev7914318019999922370
|
|
32
|
K YamaguchiK ShirakabeH ShibuyaK IrieI
OishiN UenoT TaniguchiE NishidaK MatsumotoIdentification of a
member of the MAPKKK family as a potential mediator of TGF-β signal
transductionScience2702008201119958533096
|
|
33
|
M MomcilovicSP HongM CarlsonMammalian TAK1
activates Snf1 protein kinase in yeast and phosphorylates
AMP-activated protein kinase in vitroJ Biol
Chem2812533625343200610.1074/jbc.M60439920016835226
|
|
34
|
BM EmerlingB ViolletKV TormosNS
ChandelCompound C inhibits hypoxic activation of HIF-1 independent
of AMPKFEBS
Lett58157275731200710.1016/j.febslet.2007.11.03818036344
|
|
35
|
Y GaoY ZhouA XuD WuEffects of an
AMP-activated protein kinase inhibitor, compound C, on adipogenic
differentiation of 3T3-L1 cellsBiol Pharm
Bull3117161722200810.1248/bpb.31.171618758065
|
|
36
|
DS WangK YamazakiK NohtomiK ShizumeK
OhsumiM ShibuyaH DemuraK SatoIncrease of vascular endothelial
growth factor mRNA expression by 1,25-dihydroxyvitamin D3 in human
osteoblast-like cellsJ Bone Miner
Res11472479199610.1002/jbmr.56501104088992878
|
|
37
|
HP GerberTH VuAM RyanJ KowalskiZ WerbN
FerraraVEGF couples hypertrophic cartilage remodeling, ossification
and angiogenesis during endochondral bone formationNat
Med5623628199910.1038/946710371499
|
|
38
|
JMW QuinnS TamNA SimsH SalehNE McGregorIJ
PoultonJW ScottMT GillespieBE KempBJW van DenderenGermline deletion
of AMP-activated protein kinase β subunits reduces bone mass
without altering osteoclast differentiation or functionFASEB
J242752852010
|
|
39
|
P DucyM AmlingS TakedaM PriemelAF
SchillingFT BeilJ ShenC VinsonJM RuegerG KarsentyLeptin inhibits
bone formation through a hypothalamic relay: a central control of
bone massCell100197207200010.1016/S0092-8674(00)81558-510660043
|
|
40
|
PA BaldockA SainsburyM CouzensRF
EnriquezGP ThomasEM GardinerH HerzogHypothalamic Y2 receptors
regulate bone formationJ Clin
Invest109915921200210.1172/JCI021458811927618
|
|
41
|
J CornishKE CallonU BavaC LinD NaotBL
HillAB GreyN BroomDE MyersGC NicholsonLeptin directly regulates
bone cell function in vitro and reduces bone fragility in vivoJ
Endocrinol175405415200210.1677/joe.0.175040512429038
|
|
42
|
F ElefteriouJD AhnS TakedaM StarbuckX
YangX LiuH KondoWG RichardsTW BannonM NodaLeptin regulation of bone
resorption by the sympathetic nervous system and
CARTNature434514520200510.1038/nature0339815724149
|
|
43
|
AI IdrisRJ van’t HofIR GreigSA RidgeD
BakerRA RossSH RalstonRegulation of bone mass, bone loss and
osteoclast activity by cannabinoid receptorsNat
Med11774779200510.1038/nm125515908955
|
|
44
|
G MaccarinelliV SibiliaA TorselloF
RaimondoM PittoA GiustinaC NettiD CocchiGhrelin regulates
proliferation and differentiation of osteoblastic cellsJ
Endocrinol184249256200510.1677/joe.1.0583715642801
|
|
45
|
JD AhnB DubernC Lubrano-BerthelierK
ClementG KarsentyCart overexpression is the only identifiable cause
of high bone mass in melanocortin 4 receptor
deficiencyEndocrinology14731963202200610.1210/en.2006-028116614075
|
|
46
|
S SatoR HanadaA KimuraT AbeT MatsumotoM
IwasakiH InoseT IdaM MiedaY TakeuchiCentral control of bone
remodeling by neuromedin UNat
Med1312341240200710.1038/nm164017873881
|
|
47
|
MW HamrickSL FerrariLeptin and the
sympathetic connection of fat to boneOsteoporos
Int19905912200810.1007/s00198-007-0487-917924050
|
|
48
|
LF BonewaldGR MundyRole of transforming
growth factor-β in bone remodelingClin Orthop Relat
Res2612761990
|
|
49
|
K KatoH TokudaS AdachiR
Matsushima-NishiwakiH NatsumeK YamakawaY GuT OtsukaO
KozawaAMP-activated protein kinase positively regulates
FGF-2-stimulated VEGF synthesis in osteoblastsBiochem Biophys Res
Commun400123127201010.1016/j.bbrc.2010.08.02420708602
|