Introduction
Congenital heart defects (CHDs) are the most common
developmental abnormalities with a prevalence of ~1% in neonates,
and are the leading non-infectious cause of infant mortality,
accounting for >29% of infants who die from a birth defect
(1,2). TOF is a common CHD characterized by
a gross structural abnormality of the heart with functional
significance (3) and is
complicated by ventricular septal defects, atrial septal defects or
abnormalities in the branching pattern of coronary arteries,
obstruction to right ventricular outflow tract (RVOT), aortic
dextroposition (AD) and right ventricular hypertrophy (RVH). The
abnormality of TOF starts during the first eight weeks of fetal
growth and affects ~1 in 3,000 live newborns (4). Clinical symptoms include
cyanosis/clubbing, hypoxia, breathlessness, refusal to feed,
failure to gain weight and severe congenital heart
malformation.
Classic TOF and its variants have been observed as a
heritable syndrome such as Alagille syndrome and Di Georges
syndrome. Prenatal infections, exposure to teratogens, maternal
illness and folate deficiency are the few known causes. However,
70% of TOF cases also occur sporadically, without any other
anomaly, and from unknown causes (5). The cellular and molecular mechanisms
underlying TOF are complex and involve cell coordinated growth,
specification, differentiation, migration and apoptosis,
morphogenesis and cell-cell interaction (6), and are poorly understood and
difficult to study in humans. To date, a few genes which cause TOF
have been identified. Mutations in NKX2.5 (7), GATA4 (8), TBX5 (9) and others are associated with TOF.
However, similar to most CHDs, TOF is thought to be a multigenic
disorder, and the basic mechanisms of TOF in humans are still
incompletely defined. We aimed to identify the associated causal
genes of TOF and study the pathogenesis of these genes in order to
reduce the incidence of CHDs, improving the quality of life of this
population and lowering the perinatal mortality rate.
Blood vessel epicardial substance (BVES, also known
as Popdic1 or Pop1) was discovered in 1999 by two independent
laboratories using screening to identify novel genes that were
highly expressed in the developing heart (10,11). As an evolutionarily conserved
transmembrane protein, BVES has been postulated to play a role in
cell signaling as it is localized at sites of intercellular
contacts, bound to the signaling modifier guanine nucleotide
exchange factor T, and has been shown to be involved in Rho
signaling and receptor cycling (12). Moreover, BVES has been proposed to
play a role in cell adhesion, epithelial integrity and cell
motility (13–16), three cellular functions that are
essential in embryonic gastrulation. Many studies have demonstrated
that BVES plays an important role in heart development, heart
failure and arrhythmia (17,18). Previously, we identified
differentially expressed genes between ventricular septal defect
(VSD) and normal ventricular septum myocardium using suppression
subtractive hybridization. Then, among these differentially
expressed genes, we found that the BVES gene was upregulated in the
ventricular septum of VSD patients by bioinformatics analysis
(19). We also found that Bves
expression was increased 2-fold in cardiocyte differentiation
prophase in an induced p19cl6 differentiation model. Therefore, we
hypothesized that Bves plays an important role in cardiac
development. To date, there are no reports that the BVES gene
exhibits mutations in patients with CHDs.
Herein, we sequenced the entire coding region and
splice junctions of BVES in 114 unrelated patients with TOF. The
functional characteristics of the mutant BVES were analyzed using a
luciferase reporter assay system.
Materials and methods
Samples
In the present study, blood samples were obtained
from 114 patients with TOF and 114 matched individuals with no
reported cardiac phenotype as controls who were patients
hospitalized at two medical centers in Nanjing City and Shenyang
City, respectively. In our study, the patients were diagnosed
according to their past histories, physical examination, 12-lead
electrocardiograms, and ultrasonic echocardiogram; most of the
patients had cardiac catheterization examination data and/or
operative reports. To assess the allelic frequency, another 400
unrelated healthy individuals from Peking University Hospital
received annual routine health surveys during the same period.
Patients with other congenital or acquired heart diseases were
excluded from this study. Samples of peripheral blood (2–3 ml) were
collected from all participants after ethical approval by the
Ethics Committee of Nanjing Medical University. Written informed
consent was obtained from all participants or their parents in this
human population study which conformed to the principles outlined
in the Declaration of Helsinki.
Sequencing and analysis of the purified
DNA
Human genomic DNA was isolated from
EDTA-anticoagulated blood using the proteinase K methods as
previously described (20). DNA
fragments of 200–400 bp were amplified by PCR from 10 ng of genomic
DNA from each participant using the primers listed in Table I. The amplified DNA fragments were
purified by PEG precipitation and subjected to direct sequencing on
ABI 3130XL according to the manual description of BigDye v3.1. The
gene has three transcripts, all of them coding 360 amino acid. BVES
transcript 2 that represents the variant A and variants B and C
encode the same protein. We sequenced the coding sequence (from
exon 2 to exon 8) of BVES transcript 2 (NM_007073, gi|313760594|).
Sequencing results were analyzed by SeqScanner v1.0 (Applied
Biosystems) and Mutation Surveyor v3.10 (Soft Genetics) against
references [GenBank sequence (NG_016625.1) and sequences from
controls in this study].
 | Table ISequences of primer pairs for DNA
amplification and mutational analysis. |
Table I
Sequences of primer pairs for DNA
amplification and mutational analysis.
| Exon | Forward primer
(5′-3′) | Reverse primer
(5′-3′) | Length (bp) | Annealing temperature
(°C) |
|---|
| E2-1 |
GAATCTGGGTCTGTCTAAT |
GGCACAGGTATGATACTT | 232 | 55 |
| E2-2 |
GAATCAACTGCCATAGGT |
CTCTGAAGCTCTCAATGTA | 273 | 50 |
| E3-1 |
CTGCCTGAGATGTGAATC |
CACAGAGTTCCAGATCAT | 350 | 55 |
| E3-2 |
CCGATGTGCCTTGGATAT |
CAGAAAGCCTAAACTTCAG | 226 | 50 |
| E4 |
GCTGCATTGGTGAAGCAT |
CTCCATTCATTGGCAACAT | 327 | 50 |
| E5 |
GTCCATCATCTCTGTGGAAT |
CCTTTGGATTCTGAATGAGA | 301 | 55 |
| E6-1 |
GTCCACTAAATCATTCCTAG |
GTGGGATCATTCAATGAGT | 249 | 54 |
| E6-2 |
GTCCACTAAATCATTCCTAG |
GTGGGATCATTCAATGAGT | 279 | 55 |
| E7 |
GGAATGTATGATTCAACTCC |
CCAGAGATACCCATCAGA | 250 | 55 |
| E8-1 |
GGTGCTGCTAGTGATAGAT |
GGATCTCTTCAAGACACCTT | 297 | 55 |
| E8-2 |
GTCCATCAGCTGCCTTGAT |
GCATAGTCAGAAGGCTCAGT | 242 | 55 |
| E8-3 |
GGTGTCTTGAAGAGATCCT |
GGACAACCTCATTGCTAT | 356 | 55 |
Plasmid construction and cell
transfection
Total RNA was isolated from the hearts of the
patients with hypertrophic cardiomyopathy using TRIzol reagent
(Invitrogen) according to the manufacturer's instructions. The
obtained cDNA was subjected to PCR amplification of a human
Bves encoding region with the primers:
5′-CTCCTCGAGGGCAAGCCCCTTGGAATTTT-3′ and
5′-TCTAAGCTTAAGGCAGCTGATGGACTTTC-3′. The PCR was carried out using
Pfu polymerase (Tianlab) with the following cycle profile: 3 min at
95°C followed by 35 cycles of 95°C for 30 sec, 55°C for 45 sec and
72°C for 60 sec. The resulting PCR products were then digested by
XhoI and HindIII and ligated into the pGL3-promoter
vector according to the technical manual (Promega Corp., Madison,
WI, USA) forming plasmids pGL3-Bves-WT. To create the
c.166T>C and the c.909C>T mutants, single-nucleotide point
mutations were introduced by site-directed mutagenesis using
overlap extension PCR technique. Wild-type Bves in the
pGL3-CMV-luciferase vector was used as the template forming
pGL3-exon2/7-mt. The accuracy of the plasmids was confirmed by DNA
sequencing.
We constructed plasmids that carried a CMV (human
cytomegalovirus promoter region) promoter, the human full length
coding region of BVES, and Firefly luciferase fused at its 3′-end.
The translational efficiency was measured by Firefly luciferase
activity. Transfection efficiency was standardized by reference to
Renilla luciferase activity resulting from the parallel
introduction of this plasmid into the test cells.
HEK293A cells (60–80% confluence) in 24-well plates
were transiently transfected with 0.66 μg of Bves-Firefly
luciferase fusion plasmid and 0.33 μg of PRL-TK reference
Renilla luciferase plasmid (Promega Corp.) using VigoFect
(Vigonous) according to the manufacturer's instructions.
Forty-eight hours after transfection, cells were harvested and
luciferase activity (Firefly and Renilla) was measured using
a Dual-Luciferase Reporter Assay kit (Promega Corp.) and a
multi-well fluorometer (BioTek, USA). The protein expression level
of the Bves gene was measured by the ratio of Firefly to
Renilla. Data represent the means ± SEM of twelve
independent experiments carried out in triplicate.
Statistical analysis
Sets of data in the various groups were compared
using the unpaired two-tailed t-test and expressed as the means ±
SD. A p-value of <0.05 was considered to indicate a
statistically significant result.
Results
Identification of genetic variants in
BVES
The entire Bves coding sequences of all the
subjects were analyzed and compared with the GeneBank human
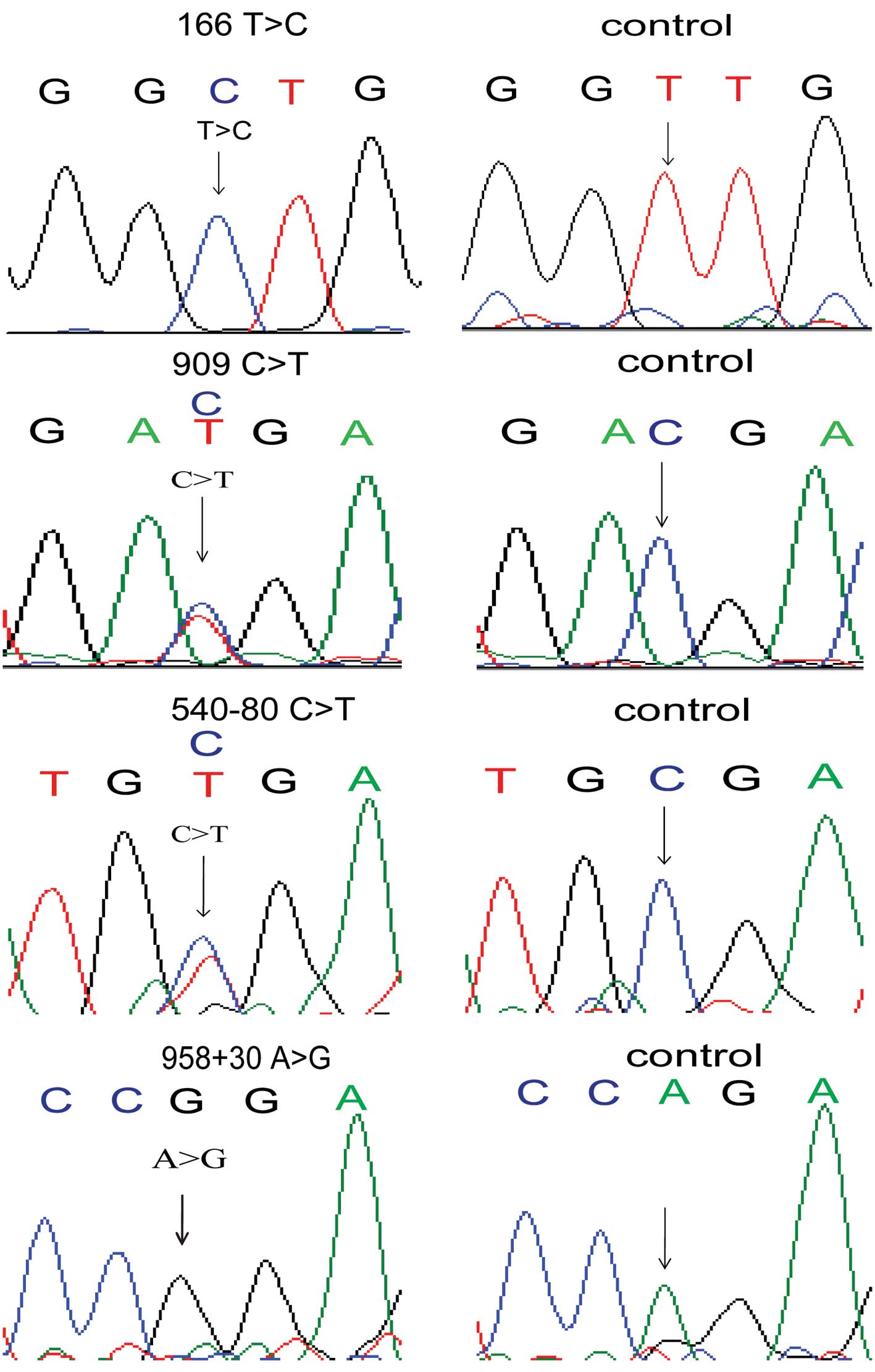
Bves coding sequence. We identified four novel varations
(c.166T>C p.L56L, c.909C>T p.D303D, c.540-80G>A,
c.958+30A>G) in four unrelated patients with TOF which were not
noted in the 114 controls nor in the additional 400 controls
(Fig. 1). In addition, we
replicated six reported single-nucleotide polymorphisms (SNPs)
(Table II). No significant
differences were found in the allelic frequencies of these SNPs
between the TOF patients and normal controls. Specifically, variant
c.166T>C at the three transmembrane helices of Bves predicted
that Bves may be lost at the cell surface. A heterozygous
(c.909C>T p.D303D) and a homozygous (c.166T>C p.L56L) variant
did not result in amino acid changes, but they may have been
impacted by the nucleotide splice or the stability of RNA. We
analyzed the nucleotide conservation of the two variants and found
that the c.166T>C variant was evolutionarily conserved (Fig. 2). To ascertain the mutations,
known TOF genes (i.e., GATA4, Nkx2.5, TBX5 and FOG2) were sequenced
and excluded from the BVES variant carriers. Clinic diagnosis of
four carriers was TOF with no clinical manifestations of
cardiomyopathies or other self-reported inherited diseases. It was
not known whether other family members of the BVES mutation
carriers had the same mutation due to a failure to obtain a signed
consent form.
| Table IIReported SNPs of the BVES gene in this
study. |
Table II
Reported SNPs of the BVES gene in this
study.
| Gene | Exon | Nucleotide | Protein | rs number | MAF | MAF reported |
|---|
| BVES | 3 | c.227-23G>A | | rs4946656 | G=14/236=0.059 | C=0.05 (90 AoD
Chinese) |
| BVES | 3 | c.351+30T>G | | rs9404604 | T=17/124=0.137 | A=0.125 (120
CHB+JPT) |
| BVES | 3 | c.351+55G>A | | rs72932419 | A=15/168=0.089 | T=0.045 (88
CHB+JPT) |
| BVES | 3 | c.351+82T>C | | rs9404603 | T=23/168=0.137 | A=0.080 (88
CHB+JPT) |
| BVES | 4 | c.385C>T | p.R129W | rs2275289 | T=4/228=0.0175 | A=0.070 (86 HCB);
0.037 (82 CHB) |
| BVES | 8 |
c.*114A>G | | rs221657 | G=26/232=0.112 | C=0.098 (86 HCB);
0.081 (82 CHB) |
Functional implications
The potential functional effects of these variants
were evaluated by bioinformatics analysis (Table III). It was found that
c.166T>C and c.958+30A>G potentially affected RNA splicing,
while c.958+30A>G produced a new silencer motif. The variant
c.540-80C>T emerged as a new enhancer motif and c.909C>T may
not be affected by RNA splicing.
| Table IIIFunctional predictions of the
variants detected in this study. |
Table III
Functional predictions of the
variants detected in this study.
| Nucleotide
change | Gene region | Wild-type | Mutant | Predicted
function |
|---|
| c.166T>C | Exon 2 | TGGGTTGGT | TGGGCTGGT | Splicing activity
decreased by −33.63% |
| c.909C>T | Exon 7 | ACAGTGACG | ACAGTGATG | Splicing activity
decreased by −0.44% |
|
c.540-80C>T/G>A | Intron 4 | AAATCG | AAATCA | New site: enhancer
motif |
| c.958+30A>G | Intron 7 | ATACTACTCCAGAG | ATACTACTCCGGAG | Splicing activity
decreased by −33.91% |
| | AGAGTT | GGAGTT | New site: silencer
motif |
Luciferase assay
To characterize the function of the mutations in
exons of BVES, mutated sequences were cloned into plasmids and
experiments were carried out compared with the wild-type control.
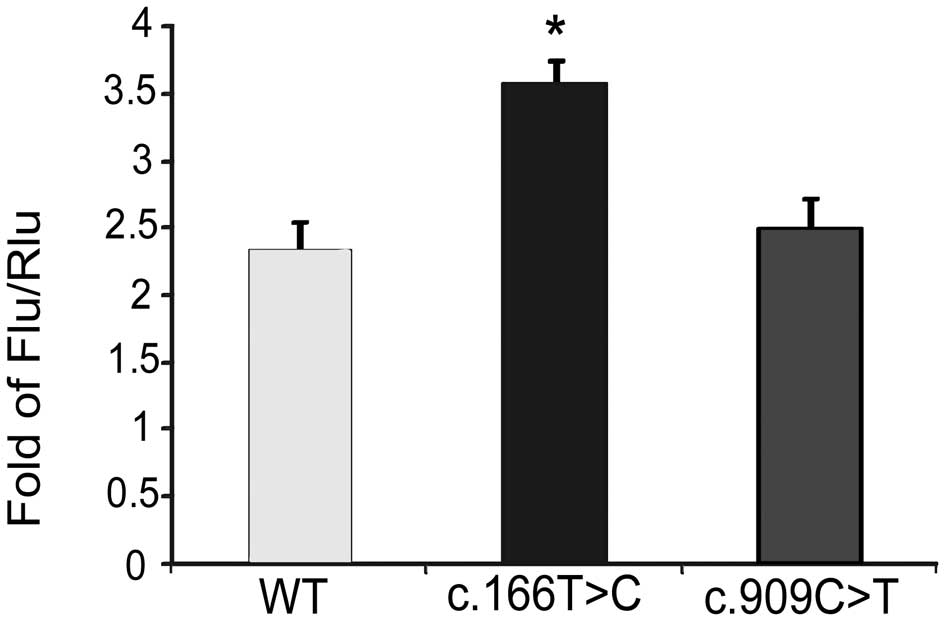
It was found that c.166T>C increased the luciferase activity by
0.5-fold (Fig. 3). Thus, we
demonstrated that the bves mutation (c.166T>C) found in TOF
patients was gain-of-function.
Discussion
The most important finding in this study was the
identification of four novel genetic variants within the BVES gene
from the peripheral blood cells of TOF patients that were not noted
in the 114 controls nor in the additional 400 controls.
Bioinformatics analysis implied that these variants potentially
affected motif-binding sites and splicing. We also demonstrated
that the bves mutation (c.166T>C) found in TOF patients
increased the luciferase activity by 0.5-fold which was
gain-of-function. There was no significant difference in the
luciferase activity between the variant c.909C>T and wild-type,
thus it was likely a rare SNP.
Tetralogy of Fallot is the most common cyanotic
congenital cardiac defect; it involves abnormalities in
myocardialization resulting in a failure to muscularize the
developing proximal outlet septum (21). TOF is of great importance to the
pediatrician and cardiac surgeon since patients with these problems
may require urgent and complicated surgery shortly after birth and
long-term follow-up is needed (5). Understanding the aetiology of these
defects would be useful for designing strategies for their
prevention, either by allowing prenatal diagnosis or by therapeutic
approaches such as vitamin supplementation that might reduce their
incidence (22).
Embryonic cardiovascular development in humans is
regulated by a diverse array of molecular signaling pathways that
are activated by various intrinsic programs, molecular and
morphogenic events (23). The
morphogenesis of the human heart is a complex process, in which
numerous genes are involved. BVES may play an important role in the
signaling pathway for heart development.
BVES is a transmembrane protein which has also been
considered as belonging to the cytoskeleton. These proteins play a
role in the link with the extracellular matrix (24). The extracellular matrix that
constitutes the cardiac jelly within the cushions has long been
thought to play an important role in early septal morphogenesis
(25). Abnormalities of the
extracellular matrix cause atrioventricular canal and outlet septum
defects (26). The differential
expression of BVES in the heart of TOF patients may cause
extracellular matrix disorder and may be involved in the
development of TOF. Our previous study found that the cytoskeleton
protein BVES was upregulated in septal defect patients (19). This study also found that the
mutation c.166T>C increased the transcription activity of BVES.
Numerous studies have demonstrated that BVES plays an important
role in heart and vascular development (27), heart failure (18) and arrhythmia (17). However, the present study was the
first to demonstrate that BVES is involved in CHD, and may be a
candidate gene of TOF.
Given the severe phenotypes noted in both X.
laevis and D. melanogastor embryos when BVES is
depleted, it was predicted that Bves-null mice would exhibit
obvious developmental defects and would not live a normal lifespan
(28). This, however, was not the
case. Bves-null mice displayed no overt morphological defects. As
the Popeye domain is highly conserved throughout all Popdc family
members, and all three members have similar tissue expression, it
is possible that Bves, Popdc2 and Popdc3 have redundant functions
in development (29). It was
demonstrated that mice lacking either Popdc1 or Popdc2 (also known
as Pop2) are normal when housed under standard conditions but
develop a striking age-dependent sinus node dysfunction when
subjected to physical or mental stress. However, Popdc1 and Popdc3
expression was not elevated in Popdc2 null mutants (17). Therefore, BVES, Popdc2 and Popdc3
do not have redundant functions in development. Previously, in
differentially expressed genes, we found that the BVES gene was
upregulated in the ventricular septum of VSD patients by
bioinformatics analysis (19).
In conclusion, we demonstrated that the BVES gene
exhibit mutations in patients with CHD, which were not previously
identified. We performed a systematic screening for genetic
mutations of the BVES gene in the peripheral blood cells of
patients with TOF and identified four novel genetic variants in TOF
cardiac tissues that were not found in the 114 controls and the
additional 400 controls. These genetic variants may be associated
with TOF through multiple mechanisms.
Acknowledgements
We thank all of the participants in our study
population. This research was supported by the National Natural
Science Fund of China (no. 30871079), the National Science
Foundation of Jiangsu (no. BK2011770) and the Medical Key Talent
Program of Jiangsu (no. K201110).
References
|
1
|
Roger VL, Go AS, Lloyd-Jones DM, et al:
American Heart Association Statistics Committee and Stroke
Statistics Subcommittee: Heart disease and stroke statistics - 2011
update: a report from the American Heart Association. Circulation.
123:e18–e209. 2011. View Article : Google Scholar
|
|
2
|
Hoffman JI: Incidence of congenital heart
disease: II. Prenata incidence. Pediatr Cardiol. 16:155–165. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mitchell SC, Korones SB, Berendes HW, et
al: Congenital heart disease in 56,109 births. Incidence and
natural history. Circulation. 43:323–332. 1971. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ferencz C, Rubin JD, McCarter RJ, et al:
Congenital heart disease: prevalence in live birth. The
Baltimore-Washington Infant Study. Am J Epidemiol. 121:31–36.
1981.
|
|
5
|
Kola S, Koneti NR, Golla JP, et al:
Mutational analysis of JAG1 gene in non-syndromic Tetralogy of
Fallot children. Clin Chim Acta. 412:2232–2236. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kaynak B, von Heydebreck A, Mebus S, et
al: Genome-wide array analysis of normal and malformed human
hearts. Circulation. 107:2467–2474. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Schott JJ, Benson DW, Basson CT, et al:
Congenital heart disease caused by mutations in the transcription
factor NKX2-5. Science. 281:108–111. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Okubo A, Miyoshi O, Baba K, Takagi M, et
al: A novel GATA4 mutation completely segregated with atrial septal
defect in a large Japanese family. J Med Genet. 41:e972004.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Basson CT, Huang T, Lin RC, et al:
Different TBX5 interactions in heart and limb defined by Holt-Oram
syndrome mutations. Proc Natl Acad Sci USA. 96:2919–2924. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Reese DE, Zavaljevski M, Streiff NL and
Bader D: bves: a novel gene expressed during coronary blood vessel
development. Dev Biol. 209:159–171. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Andree B, Hillemann T, Kessler-Icekson G,
et al: Isolation and characterization of the novel popeye gene
family expressed in skeletal muscle and heart. Dev Biol.
223:371–382. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Russ PK, Kupperman AI, Presley SH, et al:
Inhibition of RhoA signaling with increased Bves in trabecular
meshwork cells. Invest Ophthalmol Vis Sci. 51:223–230. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wilson P and Keller R: Cell rearrangement
during gastrulation of Xenopus: direct observation of
cultured explants. Development. 112:289–300. 1991.
|
|
14
|
Russ PK, Pino CJ, Williams CS, et al: Bves
modulates tight junction associated signaling. PLoS One.
6:e145632011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hager HA and Bader DM: Bves: ten years
after. Histol Histopathol. 24:777–787. 2009.PubMed/NCBI
|
|
16
|
Hager HA, Roberts RJ, Cross EE, et al:
Identification of a novel Bves function: regulation of vesicular
transport. EMBO J. 29:532–545. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Boukens BJ and Christoffels VM: Popeye
proteins: muscle for the aging sinus node. J Clin Invest.
122:810–813. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gingold-Belfer R, Bergman M, Alcalay Y, et
al: Popeye domain-containing 1 is down-regulated in failing human
hearts. Int J Mol Med. 27:25–31. 2011.
|
|
19
|
Zhang H, Zhou L, Yang R, et al:
Identification of differentially expressed genes in human heart
with ventricular septal defect using suppression subtractive
hybridization. Biochem Biophys Res Commun. 342:135–144. 2006.
View Article : Google Scholar
|
|
20
|
Tian XL and Wang QK: Generation of
transgenic mice for cardiovascular research. Methods Mol Med.
129:69–81. 2006.PubMed/NCBI
|
|
21
|
Warnes CA: The adult with congenital heart
disease: born to be bad? J Am Coll Cardiol. 46:1–8. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Giannopoulos NM, Chatzis AC, Bobos DP, et
al: Tetralogy of Fallot: influence of right ventricular outflow
tract reconstruction on late outcome. Int J Cardiol. 97(Suppl 1):
87–90. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jain R, Rentschler S and Epstein JA: Notch
and cardiac outflow tract development. Ann NY Acad Sci.
1188:184–190. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hein S, Kostin S, Heling A, et al: The
role of the cytoskeleton in heart failure. Cardiovasc Res.
45:273–278. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Maslen CL: Molecular genetics of
atrioventricular septal defects. Curr Opin Cardiol. 19:205–210.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Marino B and Digilio MC: Congenital heart
disease and genetic syndromes: specific correlation between cardiac
phenotype and genotype. Cardiovasc Pathol. 9:303–315. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wada AM, Reese DE and Bader DM: Bves:
prototype of a new class of cell adhesion molecules expressed
during coronary artery development. Development. 128:2085–2093.
2001.PubMed/NCBI
|
|
28
|
Andree B, Fleige A, Arnold HH, et al:
Mouse Pop1 is required for muscle regeneration in adult skeletal
muscle. Mol Cell Biol. 22:1504–1512. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Parnes D, Jacoby V, Sharabi A, et al: The
Popdc gene family in the rat: molecular cloning, characterization
and expression analysis in the heart and cultured cardiomyocytes.
Biochim Biophys Acta. 1769:586–592. 2007. View Article : Google Scholar : PubMed/NCBI
|