Introduction
Hepatocellular carcinoma (HCC) is one of the most
common malignancies worldwide. Liver cancer in males is the fifth
most frequently diagnosed cancer worldwide and the second most
frequent cause of cancer-related mortality (1). Half of these cases and deaths have
been estimated to occur in China. It is also the second greatest
cause of cancer-related deaths in China (2). Tumor metastasis is considered to be
the main determinant in HCC patient survival; however, the
mechanisms involved have not yet been fully elucidated. Epithelial
to mesenchymal transition (EMT) is an important step in the
invasion and metastasis of cancer and transforming growth factor-β1
(TGF-β1) induces the progression of cancer through EMT. It has been
shown that TGF-β1 plays a key role in modulating HCC aggressiveness
by triggering EMT (3). Therefore,
effective drugs targeting the TGF-β1 signaling pathways that
correlate with EMT have the potential to provide a novel approach
for the prevention and treatment of the early metastasis of HCC and
may thus improve prognosis. Histone deacetylases (HDACs) are
enzymes that catalyze the removal of acetyl from lysine residues in
histones and other proteins, causing gene transcriptional
repression and subsequent changes in signaling events. Eighteen
HDACs, which have been shown to have unique functions, have been
identified in mammals. Previous studies have demonstrated that HDAC
inhibitors suppress cancer cell invasion and metastasis by
regulating the subsequent gene transcription that causes invasion
and migration (4,5). However, it is unclear as to which
members of the HDAC family are associated with the regulation of
cancer cell invasion and migration. HDAC4, a member of class II
HDACs, has been shown to play an important role in regulating cell
migration and invasion in ovarian cancer cells (6). A high HDAC4 expression has also been
observed in liver cancer (7,8);
however, it remains unclear whether HDAC4 participates in EMT in
HCC cells.
Short chain fatty acids (SCFAs) have been reported
as effective HDAC inhibitors with multiple effects on cell growth,
differentiation and apoptosis in a variety of cancer cells
(9,10). Sodium butyrate (NaBu), one of the
sodium salts of SCFAs, is naturally produced in the colonic lumen
as a consequence of the microbial degradation of dietary fibers
(11). As an HDAC inhibitor,
first reported in 1978 (12),
NaBu has shown great potential in cancer treatment and prevention
(13). It has been demonstrated
that NaBu can cause cell cycle arrest, trigger cell apoptosis and
induce cell differentiation in multiple cancer cells lines derived
from the colon, breast, liver, etc. (14–18). Mechanism analysis has further
demonstrated that NaBu selectively regulates gene expression by
inducing chromatin hyperacetylation, which contributes to its
diverse physiological roles. The potential effects of NaBu
specifically on HCC have been investigated in a number of studies.
It has been reported that NaBu suppresses cell growth and reduces
the invasive ability of HCC cells (19,20). However, the mechanisms responsible
for these effects remain largely illusive. This prompted us to
investigate the effect of NaBu on TGF-β1-induced EMT, which may
provide a reasonable explanation for the inhibition of tumor
invasion and metastasis.
In the present study, we examined the possible role
of NaBu in the regulation of HCC cell proliferation, apoptosis,
migration and invasion. Our results show that NaBu inhibits of the
proliferation, promotes apoptosis and suppresses the invasion of
SMMC-7721 and HepG2 cells by inhibiting TGF-β1-induced EMT,
suggesting that NaBu may have therapeutic value in HCC.
Materials and methods
Cell lines and cell culture
The human HCC cell lines, HepG2, SMMC-7721, and the
L02 human normal liver cell line were purchased from the China
Center for Type Culture Collection (CCTCC). The MHCC97H and MHCC97L
cell lines were purchased from the Liver Cancer Institute of Fudan
University (Shanghai, China). The HepG2, MHCC97H, MHCC97L and L02
cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM;
Gibco), while the SMMC-7721 cells were cultured in RPMI-1640 medium
(Gibco) supplemented with 10% fetal bovine serum (FBS; Gibco), 100
U/ml of penicillin and 100 μg/ml of streptomycin in a 5%
CO2 incubator (Thermo Scientific, Barrington, IL,
USA).
Antibodies and reagents
The anti-GAPDH antibody used to evaluate the equal
loading of protein samples was purchased from Abmart, Inc.,
Shanghai, China. E-cadherin antibodies were purchased from Cell
Signaling Technology (Danvers, MA, USA). Vimentin and N-cadherin
antibody were purchased from Abcam (Cambridge, UK). p21, p27,
survivin, Bad, Bcl-2, Bcl-xL, caspase-9 and HDAC4 antibodies were
purchased from Signalway Antibody Co., Ltd., College Park, MD, USA.
Matrix metalloproteinase (MMP)7 antibody was purchased from
RayBiotech (Norcross, GA, USA). The goat anti-mouse and goat
anti-rabbit HRP-anchored secondary antibodies were purchased from
Zhongshan Goldenbridge Biotechnology, Beijing, China. NaBu was
purchased from Sigma-Aldrich (St. Louis, MO, USA), diluted with PBS
to a concentration of 100 mM and then stored at 4°C until use.
Recombinant human TGF-β1 was purchased from PeproTech (Rocky Hill,
NJ, USA), and reconstituted at 10 μg/ml in sterile PBS containing
0.1% bovine serum albumin.
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay was purchased from Fluka, Inc., USA. Annexin V-FITC apoptosis
detection kit, propidium iodide (PI) staining solution and Matrigel
were from BD Biosciences (Franklin Lakes, NJ, USA). RPMI-1640, DMEM
and FBS were from Gibco/Life Technologies, Paisley, UK.
Cell viability assay
The effect of NaBu treatment on the viability of
different cells was measured by MTT assay based on the ability of
live cells to cleave the tetrazolium ring to a molecule that
absorbs at 570 nm. Cells (1×104) 200 μl/well were grown
in 96-well plates for 24, 48 and 72 h. The cells were further
incubated with various concentrations of NaBu in DMEM or RPMI-1640
supplemented with 10% FBS. After 24, 48 and 72 h, 20 μl MTT
solution (5 mg/ml) were added to each well, and the cells were
further incubated at 37°C for 4 h; subsequently, 150 μl of buffered
DMSO were added to each well. The absorbance was recorded on a
microplate reader at a wavelength of 570 nm. Untreated cells were
used as the controls. At least 3 replicate experiments were
performed with 6 wells per concentration.
Cell cycle and apoptosis analysis by flow
cytometry
Cell cycle analyses were carried out using flow
cytometry. SMMC-7721 and HepG2 cells (1×106) were seeded
in 6-well plates and allowed to attach overnight. Growing cells
were treated with various concentrations of NaBu and harvested by
0.25% trypsin (Beyotime Institute of Biotechnology, Haimen, China)
at the indicated time points. The cells were then washed twice with
PBS, centrifuged at 1,000 rpm for 5 min, and the cells were washed
again with PBS, treated with 100 μg/ml RNase, and stained with 50
μg/ml PI (BD Biosciences, USA) in the dark. Flow cytometry analyses
were carried out on a FACScalibur instrument using the ModFit
program (Becton-Dickinson, Franklin Lakes, NJ, USA). Apoptosis was
detected with an Annexin V-FITC/PI detection kit (BD Biosciences).
Cells (10,000) per sample were analyzed with a FACScalibur flow
cytometer.
Wound healing assay
Cells were seeded into a 6-well tissue culture dish
and allowed to grow to 90% confluency in complete medium. Cell
monolayers were wounded by a plastic tip (200 μl tip) that touched
the plate as previously described (21). Wounded monolayers were then washed
3 times with medium to remove the cell debris and incubated in
medium with various concentrations of NaBu for 24, 48, 72 or 96 h,
then the distance of cell migration was determined under an
inverted microscopy at various time points. The relative migration
distance was calculated by the following formula: relative
migration distance (%) = average migration distance of treated
group/average migration distance of untreated group ×100%.
Experiments were repeated at least 3 times.
Transwell assay
The invasion assays were carried out using Transwell
chamber with 6.5 mm diameter polycarbonate filters (8 μm pore size;
Corning Costar, Rochester, NY, USA) coated with Matrigel (BD
Biosciences) in a standard manner. Cells were trypsinized and
suspended at a final concentration of 1×106 cells/ml in
DMEM or RPMI-1640 medium. Before seeding, the cells were treated
with various concentrations of NaBu for 24 h. Cell suspension was
loaded into each of the upper wells. Fetal calf serum (5%) was used
as a chemoattractant in the lower chambers. Before seeding, the
cells were incubated for 36 h at 37°C in an atmosphere of 5%
CO2/95% air, followed by a further incubation under the
same conditions; all of the non-invaded cells were then removed
from the upper face of the Transwell membrane with a cotton swab.
The invaded cells were fixed with 100% methanol and then stained
with crystal violet (Nanjing Sunshine Biotechnology, Nanjing,
China). The invaded cells were counted microscopically. Ten fields
were counted for each assay.
Reverse transcription PCR
Total RNA was isolated using TRIzol reagent
(Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s
instructions. Complementary DNA was synthesized using a reverse
transcription reagent kit (Fermentas, Burlington, ON, Canada).
Subsequently, cDNA was used to perform PCR using specific gene
primers under the condition of 95°C (3 min), followed by 35 cycles
of 94°C (30 sec), 48°C for vimentin, 52°C for N-cadherin and 58°C
for E-cadherin, GAPDH (30 sec) and 72°C (1 min), then finally 72°C
(5 min). GAPDH was employed as an endogenous control to normalize
the amount of total mRNA in each sample. The PCR products were
examined by 1.5% agarose gel electrophoresis, and the abundance of
each mRNA was detected. The reverse transcription PCR reactions
were performed in triplicate. The primer sequences were as follows:
E-cadherin sense, 5′-CGTAGCAGTGACGAATGTGGTAC-3′ and antisense,
5′-AACTGGAGAACCATTGTCTGTAGC-3′; vimentin sense,
5′-CCAAACTTTTCCTCCCTGAACC-3′ and antisense,
5′-GTGATGCTGAGAAGTTTCGTTGA-3′; N-cadherin sense,
5′-AGCCAACCTTAACTGAGGAGT-3′ and antisense,
5′-GGCAAGTTGATTGGAGGGATG-3′; GAPDH sense, 5′-ATGGGGAAGGTGAAGGTCG-3′
and antisense, 5′-GGGTCATTGATGGCAACAATATC-3′.
Western blot analysis
For the western blot analysis, cells were collected
and lysed with 1% SDS to break down the membranes, and
subsequently, whole proteins were heated at 95°C for 10 min to
inactivate the proteases. The samples were centrifuged for 15 min,
and the protein concentration of each sample was assessed using BCA
reagent (Beyotime Institute of Biotechnology). Equivalent amounts
of protein were loaded into each lane and separated using SDS-PAGE
on 10 or 12% gels (Bio-Rad, Hercules, CA, USA) and then transferred
onto PVDF membranes (Millipore, Billerica, MA, USA) by wet transfer
(Bio-Rad) and blocking with 5% non-fat milk for 1 h. Finally, the
membranes were incubated with the specific primary antibody
overnight at 4°C. After washing the membranes with Tris-buffered
saline containing Tween-20 (TBST) 3 times for 10 min each, the
membranes were incubated with HRP-conjugated secondary antibodies
for 1 h and then washed 3 times with TBST again. The target protein
bands were visualized using an ECL reagent kit (Thermo Scientific)
and exposed to an X-ray film (Kodak).
Statistical analysis
All values are expressed as the means ± SE. One-way
ANOVA followed by the LSD t-test and SNK-q test were used to
compare differences between groups. A two-sided P-value <0.05
was considered to indicate a statistically significant
difference.
Results
Effects of NaBu on Ac-H3 and HDAC4
expression in HCC cells
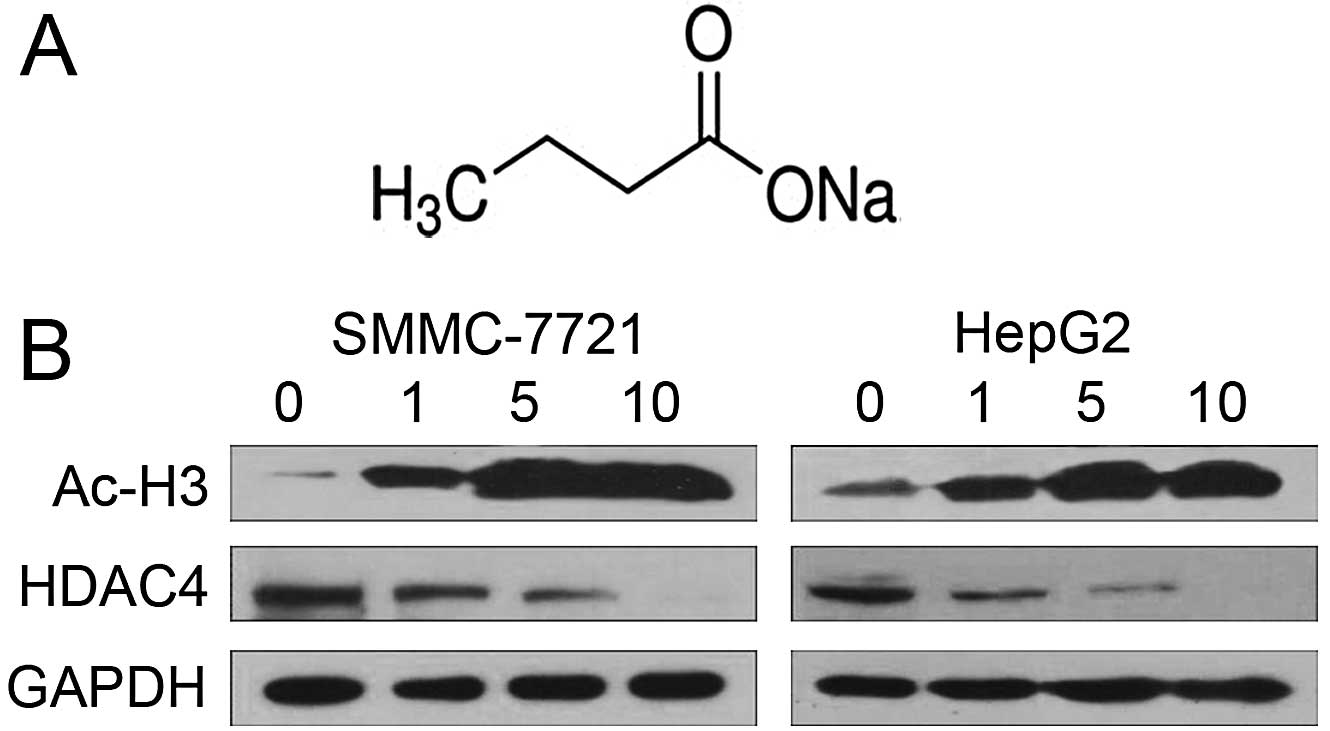
As an HDAC inhibitor, NaBu was able to induce
histone hyperacetylation. The chemical structure of NaBu is shown
in Fig. 1A. In our study, we
detected the activity of NaBu in regulating histone H3 acetylation.
SMMC-7721 and HepG2 cells were treated with various concentrations
NaBu for 24 h before being harvested and subjected to western blot
analysis using specific acetylated histone H3 (Ac-H3) antibody.
NaBu at a concentration of 0 to 10 mM (mmol/l) induced H3
acetylation in a dose-dependent manner (Fig. 1B). Western blot analysis using
specific HDAC4 antibodies was performed to further analyze the
expression of HDAC4 following NaBu treatment. The highly expressed
HDAC4 protein in SMMC-7721 and HepG2 cells was significantly
downregulated following NaBu treatment in a concentration-dependent
manner.
Different inhibitory effects of NaBu on
cell proliferation
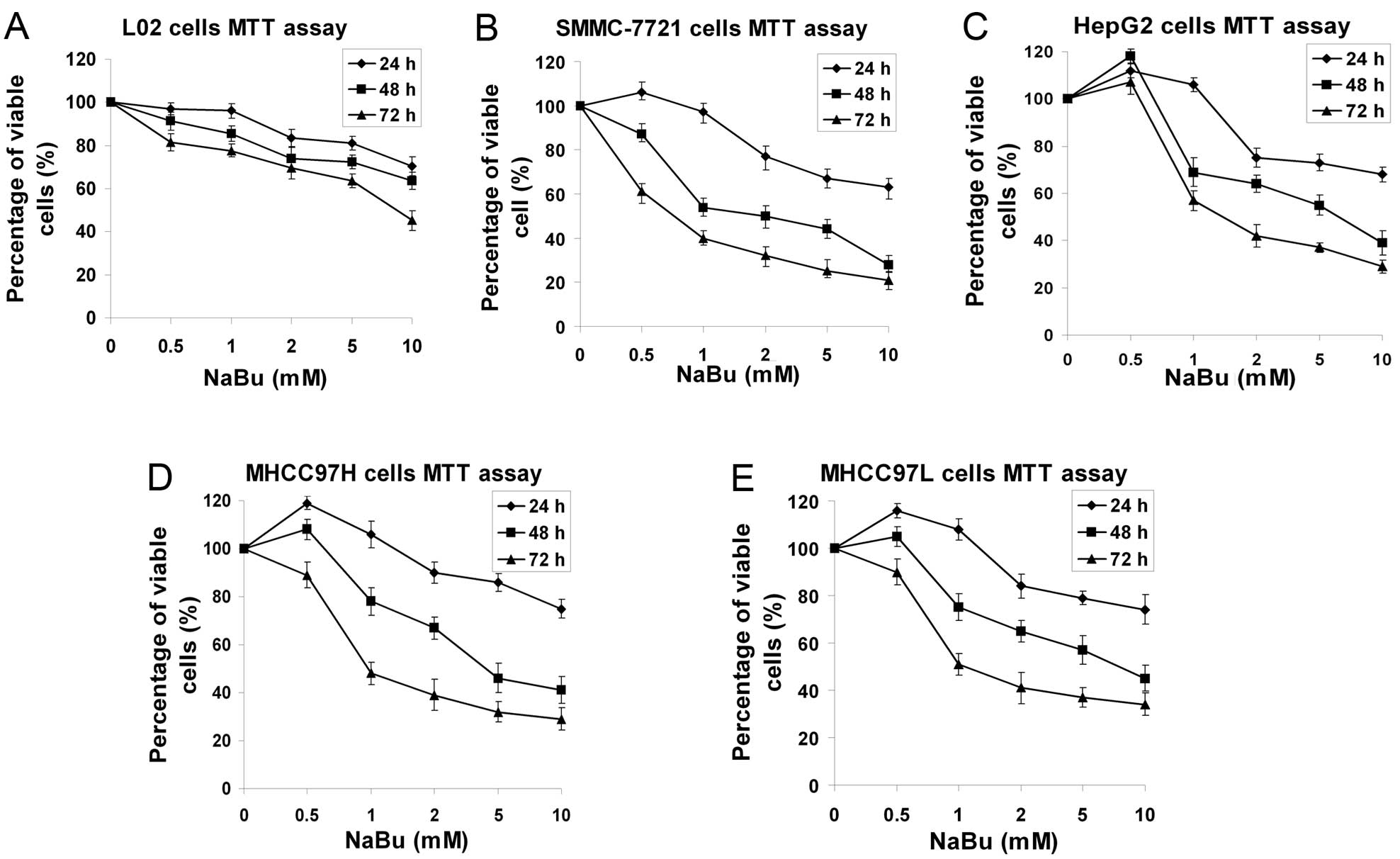
To determine the effects of NaBu on HCC cells, 4 HCC
cell lines, SMMC-7721, HepG2, MHCC97H and MHCC97L, were used in
this study. The L02 normal liver cell line was also used. All cells
were exposed to various concentrations of NaBu ranging from 0 to 10
mM for 24, 48 and 72 h, and then cell growth curves were recorded
using MTT assay. NaBu significantly inhibited the proliferation of
SMMC-7721, HepG2, MHCC97H and MHCC97L cells following treatment
with NaBu at high concentrations (Fig. 2B-E). Surprisingly, low
concentrations of NaBu (0.5 and 1 mM) had no apparent inhibitory
effect on the cells at 24 h. However, at 48 or 72 h, low
concentrations of NaBu inhibited hepatoma cell proliferation.
Furthermore, to compare these effects, the L02 normal liver cell
line was also treated with NaBu. We found that the inhibitory
effect of NaBu on L02 cells was inferior to that on HCC cells
(Fig. 2A). These data suggest
that the same concentration of NaBu exerted a stronger inhibitory
effect on hepatoma cells than L02 cells, showing low cytotoxicity
against normal liver cells at 24 h after treatment at a low dose.
These results demonstrate that low concentrations of NaBu have low
cytotoxic effects on normal liver cells in the treatment of
HCC.
Effects of NaBu on cell cycle
distribution
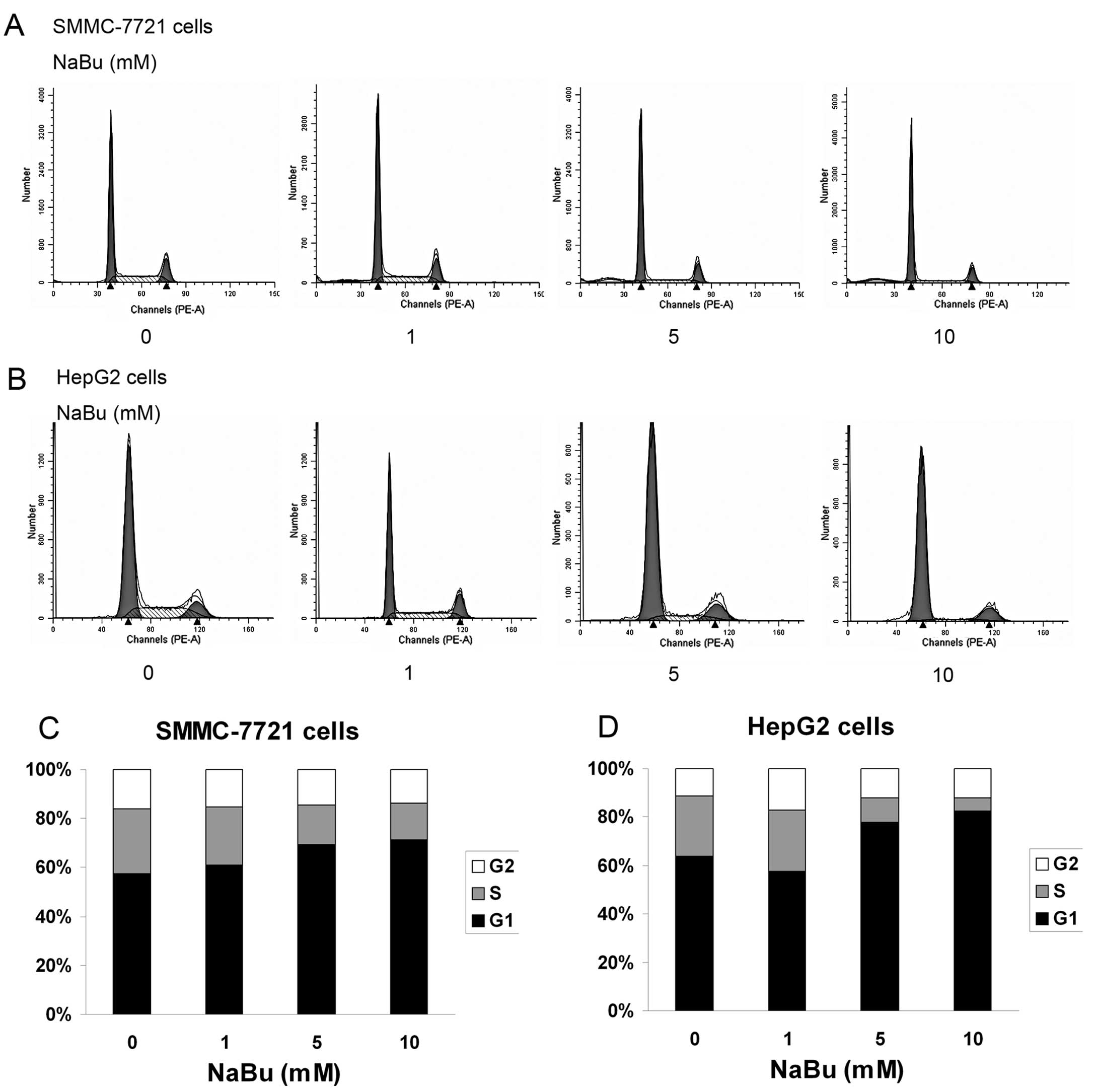
As cell cycle progression is a critical determinant
of cell growth, FACS analysis was performed to determine whether
NaBu can induce cell cycle arrest. Compared to the untreated
control groups, the SMMC-7721 and HepG2 cells treated with NaBu had
an increased percentage of cells arresting in the G1 phase at 24 h.
In addition, the S phase was significantly inhibited by treatment
with 5 and 10 mM NaBu. These data suggest that NaBu induces cell
cycle arrest in human SMMC-7721 and HepG2 cells (Fig. 3).
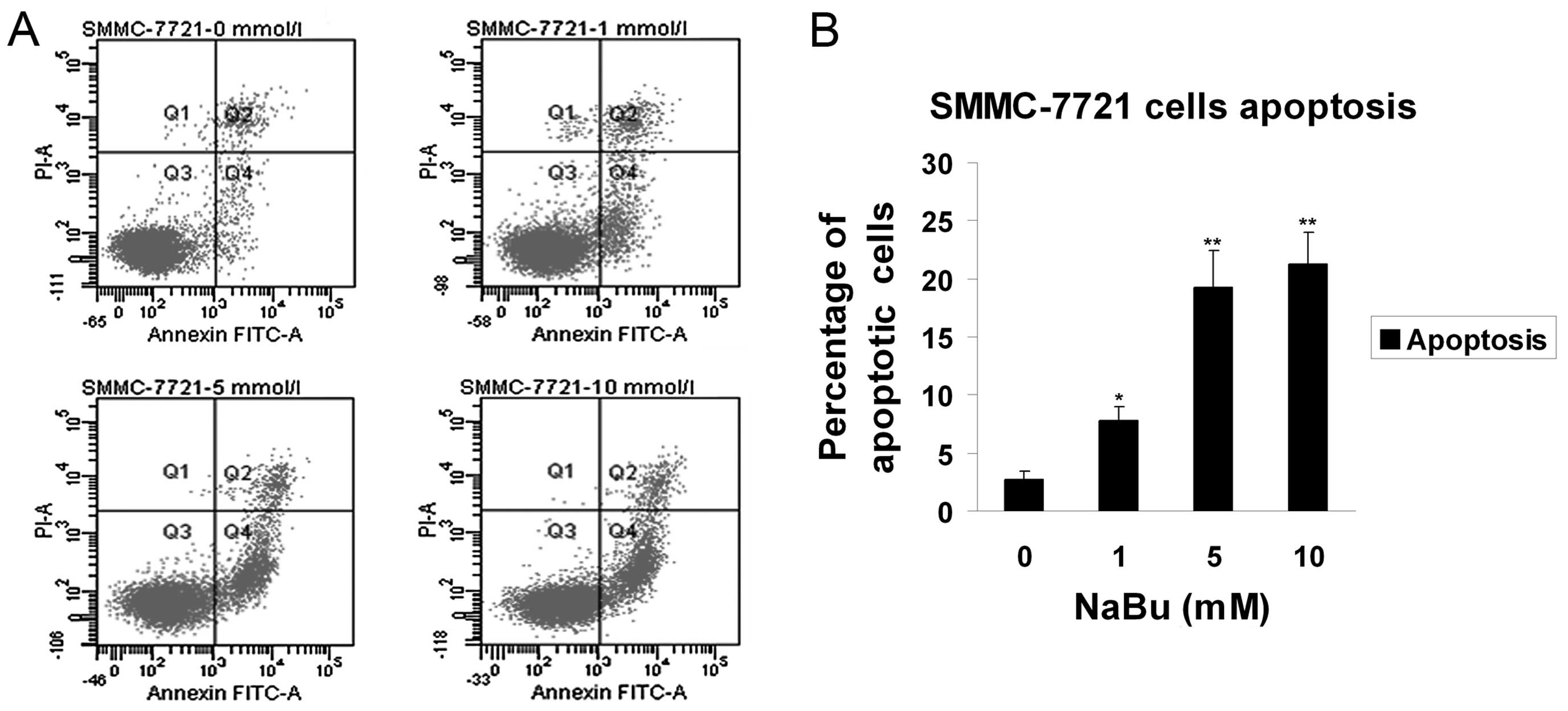
Effects of NaBu on cell apoptosis
Apoptosis induced by various concentrations of NaBu
in SMMC-7721 cells was analyzed using Annexin V/PI staining.
Compared with the control group, the proportion of apoptotic cells
increased followed by treatment with various concentrations of NaBu
(0, 1, 5 and 10 mM) for 24 h, with early apoptotic rates of 2.7,
7.8, 19.3 and 21.3%, respectively. The percentage of live cells was
93.9, 87.2, 76.1 and 75.0%, respectively (Fig. 4). These results indicated that
NaBu induced the apoptosis of SMMC-7721 cells in a concentration
and time-dependent manner. In conclusion, NaBu has the ability to
exert significant inhibitory effects on a variety of HCC cells
based on the regulation of the cell cycle and the induction of
apoptosis.
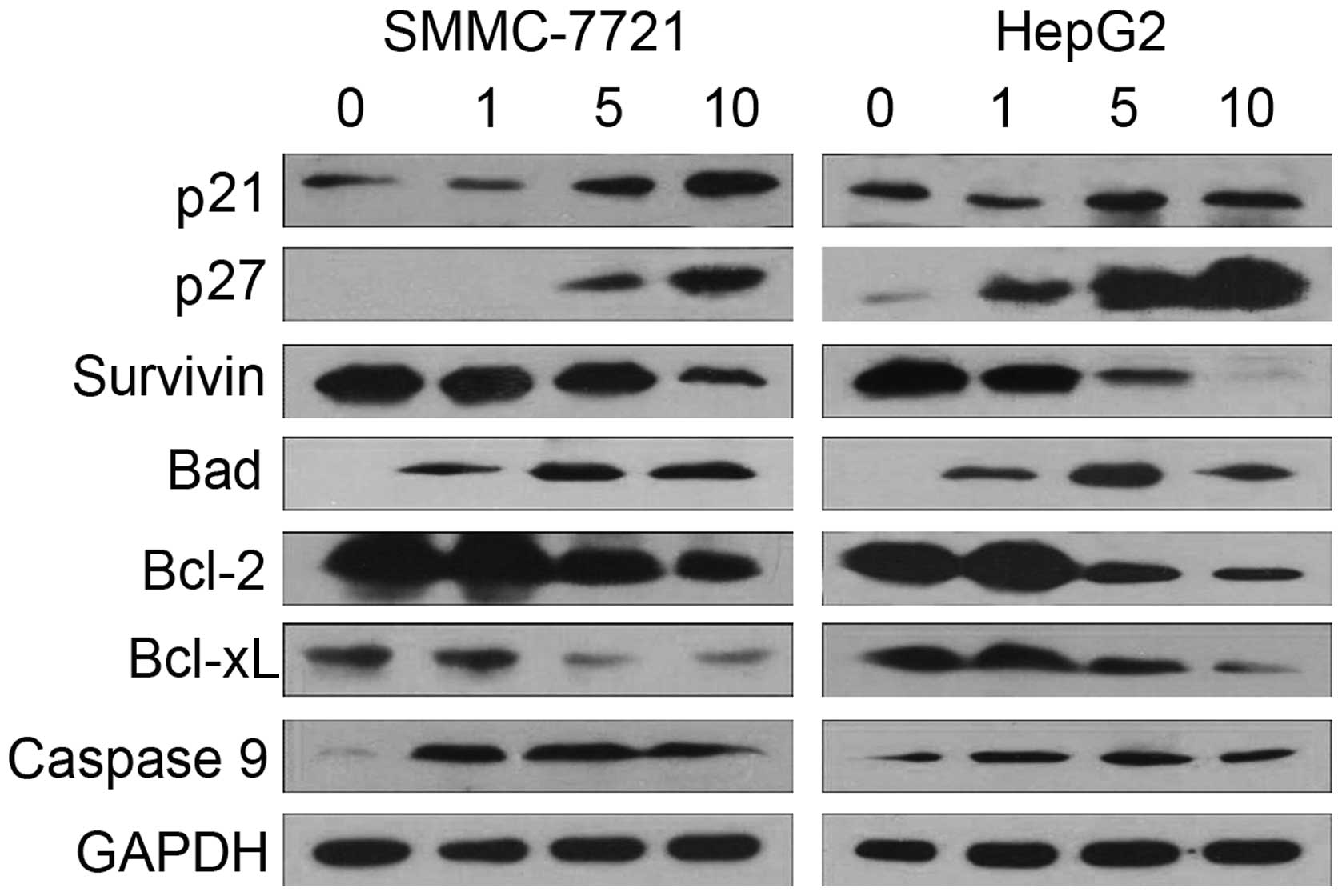
Effects of NaBu on the expression of cell
cycle- and apoptosis-related proteins
The expression levels of cell cycle- and
apoptosis-related proteins (p21, p27, survivin, Bad, Bcl-2, Bcl-xL
and caspase-9) were further investigated by western blot analysis
using specific antibodies after 24 h of NaBu treatment. As shown in
Fig. 5, high concentrations of
NaBu caused a significant increase in p21 and p27 expression;
however, 1 mM NaBu slightly reduced p21 expression. Survivin is
considered to be involved not only in cell cycle regulation, but
also in the inhibition of apoptosis. We observed a significant
reduction in survivin protein expression following treatment with
NaBu. Members of the Bcl-2 family of proteins are critical
regulators of the intrinsic apoptotic pathway. In order to
determine whether NaBu inhibits Bcl-2 family proteins, Bcl-2,
Bcl-xL and Bad expression was also detected by western blot
analysis. We found that NaBu suppressed the expression of the
anti-apoptotic proteins, Bcl-2 and Bcl-xL, in a dose-dependent
manner; however, treatment with NaBu increased pro-apoptotic
protein Bad expression in SMMC-7721 and HepG2 cells. Following NaBu
treatment, caspase-9 expression was also upregulated, suggesting
that the apoptotic process was activated in hepatoma cells. These
results provide a possible mechanism responsible for the inhibitory
effect of NaBu on the growth of HCC cells.
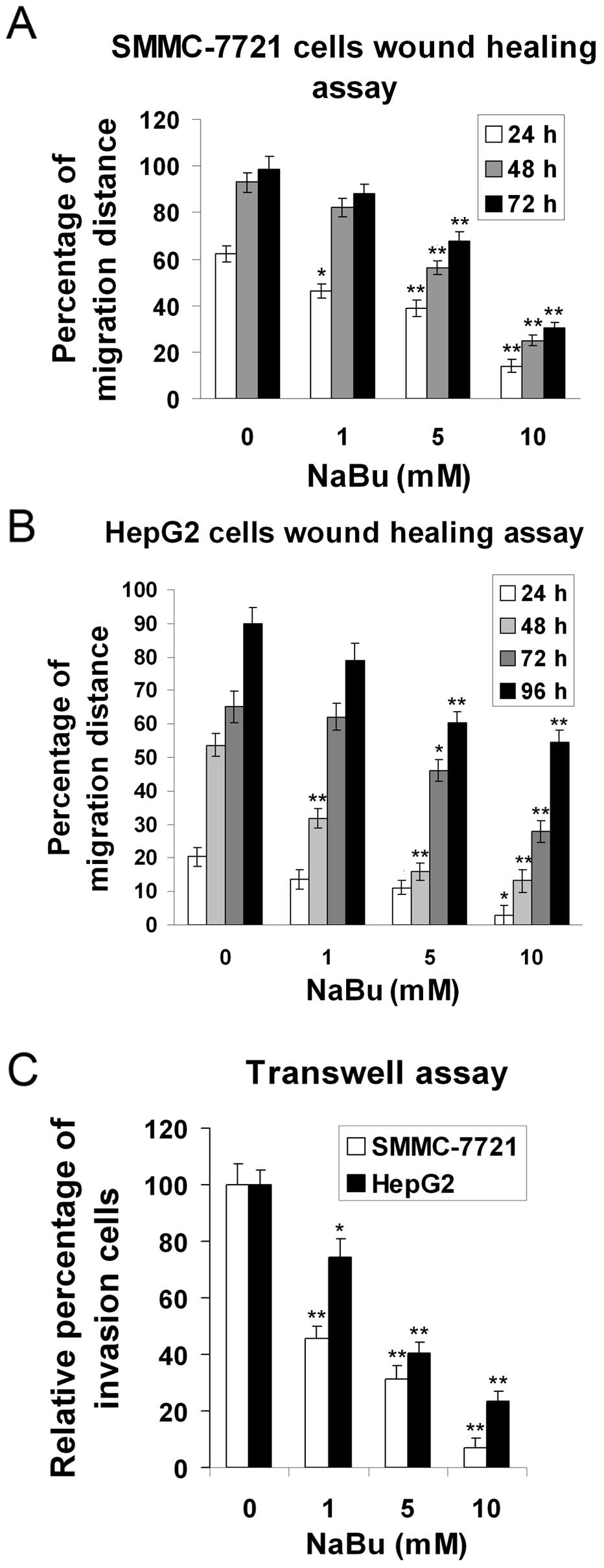
Effects of NaBu on HCC cell migration and
invasion
To further analyze the effects of NaBu on the
ability of cells to migrate and invade, wound healing and Transwell
assays were performed. The wound closure was slowed down in the
presence of NaBu in SMMC-7721 and HepG2 cells, as shown by wound
healing assay. High concentrations NaBu significantly decreased the
cell migration distance (Fig. 6A and
B). Furthermore, treatment with NaBu dramatically reduced the
invasive ability of the SMMC-7721 and HepG2 cells, as compared with
the untreated control group, as shown by Transwell assays (Fig. 6C). Taken together, these data
confer that NaBu significantly inhibits the cell migration/invasion
of HCC cells.
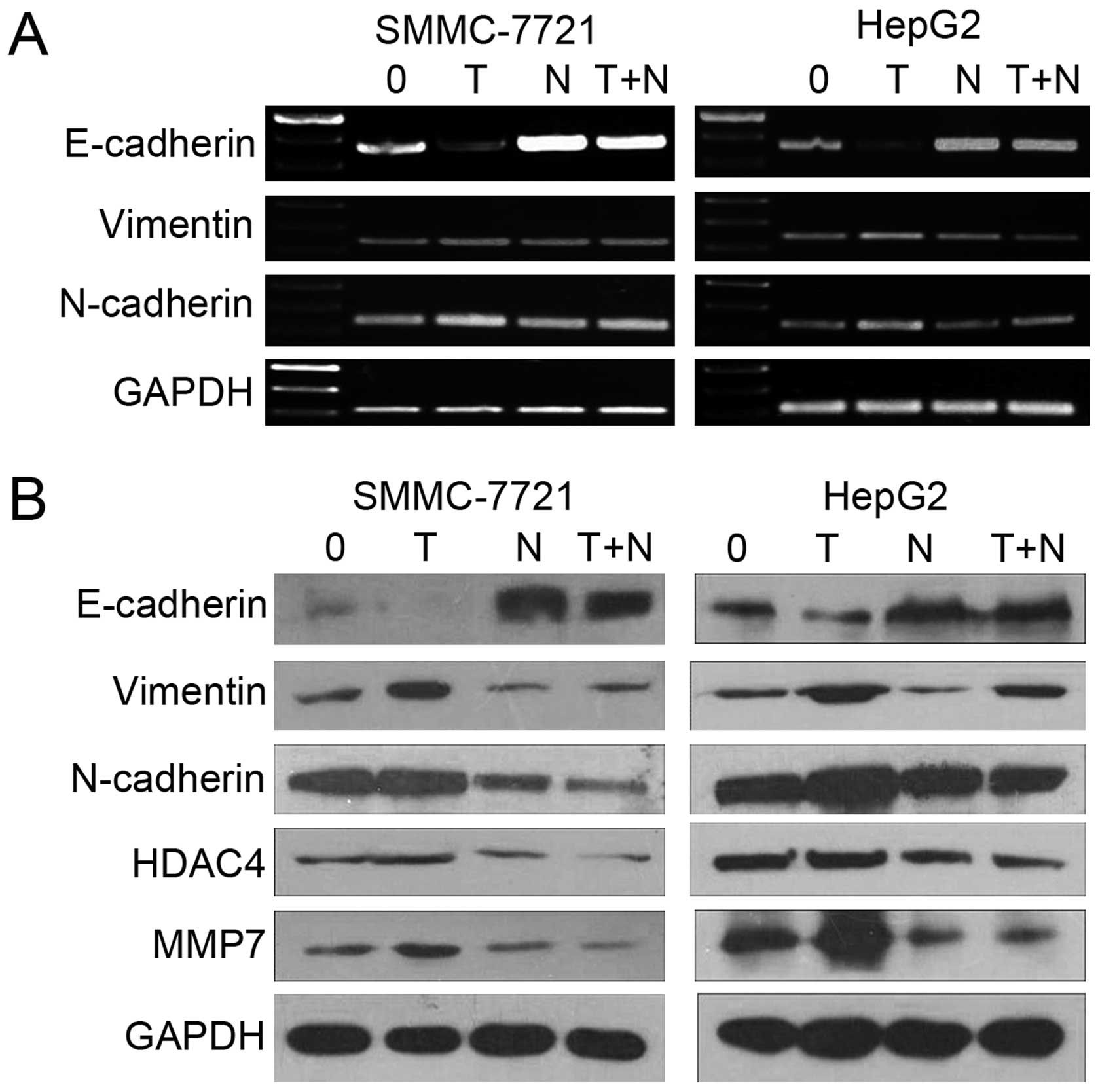
Inhibitory effects of NaBu on
TGF-β1-induced EMT
EMT has been associated with tumor cell migration
and invasion. Thus, we further examined the effects of NaBu on EMT.
Reverse transcription PCR and western blot analysis were employed
to determine whether NaBu exerts its effects by antagonizing
TGF-β1-induced EMT. E-cadherin is a universal epithelial marker in
the EMT process that plays a key role in the maintenance of
cellular integrity at the cell-cell junction. In the present study,
NaBu clearly upregulated E-cadherin mRNA and protein expression and
aborted the repression induced by TGF-β1 (Fig. 7A). Vimentin and N-cadherin are
often used to identify mesenchymal cells and the process of EMT.
MMP7, an extracellular matrix protein, is also considered be
related to EMT. Further experiments showed that NaBu aborted the
TGF-β1-induced upregulation of vimentin, N-cadherin, MMP7 and HDAC4
in the SMMC-7721 and HepG2 cells (Fig. 7B). Taken together, these data
suggest that NaBu inhibits TGF-β1-induced EMT in human hepatoma
cells, suggesting that NaBu exerts an inhibitory effect on the
early metastasis HCC cells.
Discussion
The modification of epigenetic markers, such as DNA
methylation or histone acetylation by pharmacological agents has
been hypothesized to play a role in cancer progression (22). Epigenetic regulation of gene
transcription by histone acetylation has been shown to be a
promising new therapeutic target in oncology (22–24). Different patterns of HDAC
overexpression have been demonstrated for a variety of human
cancers (25–28). HDAC4, a class II HDAC, has been
associated with a number of diseases, such as neurodegeneration
(29), diabetes (30) and cancer process (6,31).
A recent study showed that HDAC4 mRNA was upregulated in human HCC
tissues compared with paired adjacent non-cancerous hepatic
tissues, as analyzed by real-time PCR (7), suggesting HDAC4 plays a role in HCC
progression. An effective inhibitor of HDAC4 may therefore prove
useful in the treatment of HCC. NaBu, a HDAC inhibitor, inhibits
the majority of HDACs, apart from class III HDACs and the class II
HDACs, HDAC6 and HDAC10 (16). In
the current study, we observed that NaBu downregulated HDAC4
expression in an dose-dependent manner in the SMMC-7721 and HepG2
cells, proving NaBu is a potent inhibitor of HDAC4 in HCC
cells.
As a HDAC inhibitor, NaBu affects cell behavior
through the transcriptional regulation of gene expression.
Epigenetic markers, such as histone acetylation and chromatin
plasticity may be modified upon NaBu treatment. In present study,
NaBu induced histone H3 acetylation in a dose-dependent manner.
This was observed in the HepG2 cells, providing a possible
mechanism for gene expression regulation, as described above. We
then analyzed the effect of NaBu on the L02 normal liver cell line
and on 4 HCC cell lines by MTT. We found that NaBu exerted a
significant inhibitory effect on the HCC cell lines treated with
high concentrations of NaBu. However, no potent inhibitory effect
was observed in the normal liver cell line following treatment with
low concentrations of NaBu. This indicates that NaBu may exert
weaker cytotoxic effects on normal liver cells compared to HCC
cells.
We demonstrated that NaBu is highly effective in
suppressing the growth of HCC cells. The arrest in the G1 phase of
the cell cycle and the reduction in the S phase cell population is
likely to account for this effect. The molecular mechanism involved
was further examined by western blot analysis. p21 and p27 are
cyclin-dependent kinase inhibitors that play important roles in
blocking the cell cycle in the G1 phase (32). The protein levels of p21 and p27
were upregulated following treatment with NaBu at high
concentrations, suggesting that this upregulation is one of the
possible mechanisms by which NaBu inhibits the growth of HCC cells.
Apoptosis is an active gene-directed mechanism of cellular suicide,
important for the development and homeostasis of the multicellular
organism. Specific therapies have been designed to enhance the
susceptibility of human cancer cells to undergo apoptosis. In this
study, we showed that NaBu dramatically and significantly increased
the number of early apoptotic cells in the SMMC-7721 cell line.
This effect was associated with a decrease in the levels of the
anti-apoptotic proteins, survivin, Bcl-2, Bcl-xL and an increase in
the levels of the pro-apoptotic proteins, Bad and caspase-9. The
present findings raise the possibility that NaBu may prove
particularly effective in the treatment of HCC. Thus, NaBu exhibits
anti-proliferative activity and potently induces apoptosis in human
liver cancer cells.
Tumor invasion and metastasis are the major
characteristics of aggressive phenotypes of a variety of human
cancers, and, therefore, the major causes of cancer-related
mortality. HDAC inhibitors have been shown to be inhibitors of
migration and invasion (6,33).
In this study, wound healing and Transwell assays showed that a
significant decrease in HCC cell migration and invasion occurred
following treatment with NaBu at various concentrations. However,
the detailed mechanisms involved remain unclear. EMT has been
associated with tumor invasion and metastasis (34,35). TGF-β1 can induce typical EMT in a
number of cells, including hepatocytes and HCC cells, which has
been implicated in hepatic fibrosis, cirrhosis, and HCC invasion
and metastasis (36–39). It has been reported that another
HDAC inhibitor, trichostatin A (TSA), suppresses the TGF-β1-induced
EMT in hepatocytes (40) and
renal epithelial cells (41).
However, it remains unclear whether NaBu participates in
TGF-β1-induced EMT in HCC cells. Thus, we further detected
EMT-related gene expression following treatment with 10 ng/ml
TGF-β1 and/or 1 mM NaBu. We observed no significant cytotoxic
effects on normal liver cells treated with 1 mM NaBu for 24 h. In
the present study, we found that NaBu abolished the downregulation
of E-cadherin mRNA and protein expression induced by TGF-β1 in
SMMC-7721 and HepG2 cells. The mesenchymal markers, N-cadherin and
vimentin, were upregulated by TGF-β1; however, NaBu suppressed this
process. Taken together, these results demonstrate that NaBu
prevented TGF-β1-induced EMT, thus providing a possible mechanism
responsible for its inhibitory effect on migration and invasion. A
recent study (6) revealed that
apicidin, a HDAC inhibitor, inhibited human ovarian cancer cell
migration via HDAC4 silencing, showing that HDAC4 may be involved
in EMT in cancer. In this study, the HDAC4 inhibitor, NaBu,
inhibited TGF-β1-induced EMT; therefore, we propose that HDAC4 may
also participate in TGF-β1-induced EMT. In our study, we found that
HDAC4 levels were elevated following treatment with TGF-β1. The
upregulated HDAC4 expression was suppressed following treatment
with NaBu in the SMMC-7721 and HepG2 cells. It has been
substantiated that the TGF-β1 signaling pathway can activate MMP7
expression (42–44) which is able to cleave and shed
digestive E-cadherin (42). In
our study, we found that NaBu suppressed the TGF-β1-induced
upregulation of MMP7 expression in both HCC cells, providing a new
mechanism responsible for the inhibitory effect of NaBu on
TGF-β1-induced EMT. Taken together, our results showed that NaBu
suppressed cell migration and invasion by inhibiting TGF-β1-induced
EMT, in which HDAC4 and MMP7 may be involved.
In conclusion, our study demonstrates the inhibitory
effects of the HDAC inhibitor, NaBu. These effects include the
inhibition of cell proliferation, cell cycle arrest, the regulation
of TGF-β1-induced EMT and the inhibition of cell migration and
invasion in HCC cells. These data suggest that such activities
serve as anti-tumor mechanisms of the HDAC inhibitor, NaBu, in
which HDAC4 and MMP7 may be involved. However, further research is
required. In future studies, we aim to investigate the role of NaBu
in animals to verify whether there is a similar anticancer effect.
In conclusion, NaBu exerts anticancer effects on HCC cells in
vitro; thus, NaBu may provide a novel strategy for the
treatment of HCC.
Acknowledgements
This study was supported by the Science and
Technology Program of the Health Department of Jiangsu Province
(H201009), the Science and Technology Innovation Foundation of
Nanjing Medical University (2010NJMUZ054) and the Program of
Science and Technology of Nanjing (201104029). The funding sources
had input into the design of this study, the collection, analysis
and interpretation of the data.
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar
|
|
2
|
Tang ZY: Small hepatocellular carcinoma:
current status and prospects. Hepatobiliary Pancreat Dis Int.
1:349–353. 2002.PubMed/NCBI
|
|
3
|
Giannelli G, Bergamini C, Fransvea E,
Sgarra C and Antonaci S: Laminin-5 with transforming growth
factor-beta1 induces epithelial to mesenchymal transition in
hepatocellular carcinoma. Gastroenterology. 129:1375–1383. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Marks PA, Richon VM, Miller T and Kelly
WK: Histone deacetylase inhibitors. Adv Cancer Res. 91:137–168.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jeon HW and Lee YM: Inhibition of histone
deacetylase attenuates hypoxia-induced migration and invasion of
cancer cells via the restoration of RECK expression. Mol Cancer
Ther. 9:1361–1370. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ahn MY, Kang DO, Na YJ, et al: Histone
deacetylase inhibitor, apicidin, inhibits human ovarian cancer cell
migration via class II histone deacetylase 4 silencing. Cancer
Lett. 325:189–199. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yuan JH, Yang F, Chen BF, et al: The
histone deacetylase 4/SP1/microrna-200a regulatory network
contributes to aberrant histone acetylation in hepatocellular
carcinoma. Hepatology. 54:2025–2035. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang J, Yang Y, Yang T, et al:
microRNA-22, downregulated in hepatocellular carcinoma and
correlated with prognosis, suppresses cell proliferation and
tumourigenicity. Br J Cancer. 103:1215–1220. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Prasad KN: Butyric acid: a small fatty
acid with diverse biological functions. Life Sci. 27:1351–1358.
1980. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
D’Argenio G and Mazzacca G: Short-chain
fatty acid in the human colon. Relation to inflammatory bowel
diseases and colon cancer. Adv Exp Med Biol. 472:149–158.
1999.PubMed/NCBI
|
|
11
|
Bergman EN: Energy contributions of
volatile fatty acids from the gastrointestinal tract in various
species. Physiol Rev. 70:567–590. 1990.PubMed/NCBI
|
|
12
|
Candido EP, Reeves R and Davie JR: Sodium
butyrate inhibits histone deacetylation in cultured cells. Cell.
14:105–113. 1978. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fung KY, Cosgrove L, Lockett T, Head R and
Topping DL: A review of the potential mechanisms for the lowering
of colorectal oncogenesis by butyrate. Br J Nutr. 108:820–831.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Archer SY, Meng S, Shei A and Hodin RA:
p21(WAF1) is required for butyrate-mediated growth inhibition of
human colon cancer cells. Proc Natl Acad Sci USA. 95:6791–6796.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Fung KY, Brierley GV, Henderson S, et al:
Butyrate-induced apoptosis in HCT116 colorectal cancer cells
includes induction of a cell stress response. J Proteome Res.
10:1860–1869. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Davie JR: Inhibition of histone
deacetylase activity by butyrate. J Nutr. 133(Suppl 7):
2485S–2493S. 2003.PubMed/NCBI
|
|
17
|
Sun B, Liu R, Xiao ZD and Zhu X: c-MET
protects breast cancer cells from apoptosis induced by sodium
butyrate. PLoS One. 7:e301432012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tsutsumi T, Ido A, Nakao K, et al:
Reciprocal regulation of alpha-fetoprotein and albumin gene
expression by butyrate in human hepatoma cells. Gastroenterology.
107:499–504. 1994.PubMed/NCBI
|
|
19
|
Yamamoto H, Fujimoto J, Okamoto E,
Furuyama J, Tamaoki T and Hashimoto-Tamaoki T: Suppression of
growth of hepatocellular carcinoma by sodium butyrate in vitro and
in vivo. Int J Cancer. 76:897–902. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kaneko F, Saito H, Saito Y, et al:
Down-regulation of matrix-invasive potential of human liver cancer
cells by type I interferon and a histone deacetylase inhibitor
sodium butyrate. Int J Oncol. 24:837–845. 2004.PubMed/NCBI
|
|
21
|
Lok GT, Chan DW, Liu VW, et al: Aberrant
activation of ERK/FOXM1 signaling cascade triggers the cell
migration/invasion in ovarian cancer cells. PLoS One. 6:e237902011.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dawson MA and Kouzarides T: Cancer
epigenetics: from mechanism to therapy. Cell. 150:12–27. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Perego P, Zuco V, Gatti L and Zunino F:
Sensitization of tumor cells by targeting histone deacetylases.
Biochem Pharmacol. 83:987–994. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ganesan A, Nolan L, Crabb SJ and Packham
G: Epigenetic therapy: histone acetylation, DNA methylation and
anti-cancer drug discovery. Curr Cancer Drug Targets. 9:963–981.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Quint K, Agaimy A, Di Fazio P, et al:
Clinical significance of histone deacetylases 1, 2, 3, and 7: HDAC2
is an independent predictor of survival in HCC. Virchows Arch.
459:129–139. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Francisco R, Perez-Perarnau A, Cortes C,
Gil J, Tauler A and Ambrosio S: Histone deacetylase inhibition
induces apoptosis and autophagy in human neuroblastoma cells.
Cancer Lett. 318:42–52. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang JX, Li DQ, He AR, et al: Synergistic
inhibition of hepatocellular carcinoma growth by cotargeting
chromatin modifying enzymes and poly (ADP-ribose) polymerases.
Hepatology. 55:1840–1851. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang J, Kim TH, Ahn MY, et al: Sirtinol, a
class III HDAC inhibitor, induces apoptotic and autophagic cell
death in MCF-7 human breast cancer cells. Int J Oncol.
41:1101–1109. 2012.PubMed/NCBI
|
|
29
|
Li J, Chen J, Ricupero CL, et al: Nuclear
accumulation of HDAC4 in ATM deficiency promotes neurodegeneration
in ataxia telangiectasia. Nat Med. 18:783–790. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Weems JC, Griesel BA and Olson AL: Class
II histone deacetylases downregulate GLUT4 transcription in
response to increased cAMP signaling in cultured adipocytes and
fasting mice. Diabetes. 61:1404–1414. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Reddy SD, Pakala SB, Molli PR, et al:
Metastasis-associated protein 1/histone deacetylase 4-nucleosome
remodeling and deacetylase complex regulates phosphatase and tensin
homolog gene expression and function. J Biol Chem. 287:27843–27850.
2012. View Article : Google Scholar
|
|
32
|
Izutani Y, Yogosawa S, Sowa Y and Sakai T:
Brassinin induces G1 phase arrest through increase of p21 and p27
by inhibition of the phosphatidylinositol 3-kinase signaling
pathway in human colon cancer cells. Int J Oncol. 40:816–824.
2012.PubMed/NCBI
|
|
33
|
Catalano MG, Fortunati N, Pugliese M, et
al: Histone deacetylase inhibition modulates E-cadherin expression
and suppresses migration and invasion of anaplastic thyroid cancer
cells. J Clin Endocrinol Metab. 97:E1150–E1159. 2012. View Article : Google Scholar
|
|
34
|
Thiery JP, Acloque H, Huang RY and Nieto
MA: Epithelial-mesenchymal transitions in development and disease.
Cell. 139:871–890. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yang J and Weinberg RA:
Epithelial-mesenchymal transition: at the crossroads of development
and tumor metastasis. Dev Cell. 14:818–829. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Iredale JP: Models of liver fibrosis:
exploring the dynamic nature of inflammation and repair in a solid
organ. J Clin Invest. 117:539–548. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zeisberg M, Yang C, Martino M, et al:
Fibroblasts derive from hepatocytes in liver fibrosis via
epithelial to mesenchymal transition. J Biol Chem. 282:23337–23347.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Nitta T, Kim JS, Mohuczy D and Behrns KE:
Murine cirrhosis induces hepatocyte epithelial mesenchymal
transition and alterations in survival signaling pathways.
Hepatology. 48:909–919. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Fransvea E, Angelotti U, Antonaci S and
Giannelli G: Blocking transforming growth factor-beta up-regulates
E-cadherin and reduces migration and invasion of hepatocellular
carcinoma cells. Hepatology. 47:1557–1566. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kaimori A, Potter JJ, Choti M, Ding Z,
Mezey E and Koteish AA: Histone deacetylase inhibition suppresses
the transforming growth factor beta1-induced
epithelial-to-mesenchymal transition in hepatocytes. Hepatology.
52:1033–1045. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Yoshikawa M, Hishikawa K, Marumo T and
Fujita T: Inhibition of histone deacetylase activity suppresses
epithelial-to-mesenchymal transition induced by TGF-beta1 in human
renal epithelial cells. J Am Soc Nephrol. 18:58–65. 2007.
View Article : Google Scholar
|
|
42
|
Cavallaro U and Christofori G: Cell
adhesion and signalling by cadherins and Ig-CAMs in cancer. Nat Rev
Cancer. 4:118–132. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
43
|
Jovanovic V, Dugast AS, Heslan JM, et al:
Implication of matrix metalloproteinase 7 and the noncanonical
wingless-type signaling pathway in a model of kidney allograft
tolerance induced by the administration of anti-donor class II
antibodies. J Immunol. 180:1317–1325. 2008. View Article : Google Scholar
|
|
44
|
Osteen KG, Keller NR, Feltus FA and Melner
MH: Paracrine regulation of matrix metalloproteinase expression in
the normal human endometrium. Gynecol Obstet Invest. 48(Suppl 1):
S2–S13. 1999. View Article : Google Scholar
|