Introduction
WWOX (WW domain containing oxidoreductase) is a
tumor-suppressor gene located at 16q23.3–24.1, a chromosome region
that spans the second most common human fragile site named FRA16D
(1). The WWOX gene is composed of
nine exons and encodes a 46-kDa Wwox protein that consists of two
N-terminal WW domains and one C-terminal short-chain dehydrogenase
domain named SDR (1–3). WW domains are characterized for
their interactions with proline-containing ligands, and they play
an important role in mediating protein-protein interactions
(3). The SDR domain is located in
the central region of Wwox, and contains amino acid sequence
homology to the steroid oxidoreductases (3–5).
It has been demonstrated that WWOX plays a functional role as a
tumor-suppressor gene, as loss or alteration of WWOX was found in
multiple types of solid cancers including breast, lung, esophagus,
pancreas, and other cancers (6–10).
When overexpressed, WWOX is capable of initiating apoptosis in
vitro and suppresses tumor growth in vivo (11,12). WWOX-knockout mice also showed a
shortened lifespan or defects in bone metabolism or increased
incidence of tumorigenesis, as well as other deficiencies (11,13).
WWOX partners consist of an extensive scope
including the apoptosis-associated factors p73 and p53 (14–18). Studies have reported that ectopic
WWOX exhibits proapoptotic and tumor inhibitory functions, probably
by interacting with p73 or p53 (17), and WWOX expression triggers
redistribution of p73 from the nucleus to the cytoplasm. In
addition, the proapoptotic activity of WWOX can be enhanced by
cytoplasmic p73 (14). Gomes
et al recently found that WWOX mRNA levels are
associated with p53 (18).
Therefore, whether WWOX interacts with p73 or p53 in humans is not
well validated. Thus, we investigated the effects of WWOX
overexpression on the biological properties of Jurkat and K562
cells, and observed a dramatic growth arrest in these cell lines.
To elucidate the underlying mechanisms, we investigated whether
WWOX interacts with p73 or p53. We initially increased WWOX
expression in Jurkat (WWOX mRNA and Wwox absent) and K562
(WWOX mRNA low expression and Wwox absent) cells by
transfecting them with the pGC-FU-WWOX lentiviral plasmid, and then
traced the interactions between WWOX and p73 or p53 via
co-immunoprecipitation and western blot assays. We confirmed
specific interactions between WWOX and p73 in human leukemia.
Materials and methods
Materials
Jurkat and K562 cells were purchased from the
Chinese Academy of Sciences (Shanghai, China). The main reagents
are listed as follows: RPMI-1640, FBS (Gibco-BRL, Carlsbad, CA,
USA); TRIzol reagent, Lipofectamine 2000 (Invitrogen, USA);
AgeI enzyme (NEB, USA); RT-PCR kit (Fermentas, USA); qPCR
kit (Roche Diagonostics, USA); pGC-FU lentiviral vector, pHelper
1.0, pHelper 2.0, T4 DNA ligase (Genechem, Shanghai, China); rabbit
anti-human Wwox, mouse monoclonal anti-human p53, anti-p73,
anti-lamin B (Abcam, USA); RIPA lysis buffer, Co-IP kit, DAPI,
mouse anti-human β-actin, FITC-conjugated anti-rabbit IgG,
Cy3-conjugated anti-mouse IgG, mouse anti-human tubulin (Beyotime,
Shanghai, China); nuclear and cytoplasmic protein extraction kit
(Zoman Biotechnology, Beijing, China).
Cell culture
Jurkat and K562 cells were maintained in RPMI-1640
supplemented with 10% fetal bovine serum, and cultured at 37°C in
5% CO2.
Construction of the WWOX lentiviral
vectors
The human WWOX gene was cloned from normal human
liver via RT-PCR with the primer pairs: F, 5′-GAGGATCCCCGG
GTACCGGTCGCCACCATGGCAGCGCTGCGCTAC-3′; R,
5′-TCACCATGGTGGCGACCGGGCCGGACTGGCTGCC AAG-3′. The PCR product was
digested by AgeI enzyme, and the lentiviral vector (pGC-FU)
was treated in the same manner. WWOX cDNA and pGC-FU were
integrated with the aid of the T4 DNA ligase, followed by
transformation, clone picking and amplification, respectively. The
combined plasmids were extracted and sent for sequencing. Two types
of package vectors (pHelper 1.0 and pHelper 2.0) were required to
make the pGC-FU recombinant integrated. All vectors were propagated
in 293T cells using Lipofectamine 2000.
Cell infection and cell growth
assays
pGC-FU-WWOX (encoding Wwox-GFP fusion protein),
pGC-FU-GFP (a mock plasmid only encoding GFP) and untreated cells
(blank control) were established. Cells were seeded at a density of
1×105/ml, and the lentiviral plasmid was added with an
optimal MOI of 50 for Jurkat and MOI of 30 for K562 cells. A
commercial Cell Counting Kit-8 (Dojindo, Japan) was used to
evaluate the growth-inhibition effects according to the specified
protocol. The optical density (OD value proportional to the cell
number) was measured with a microculture plate reader (Bio Tek
Instruments, USA) at both 450 and 630 nm.
Reverse transcription-PCR analysis
Total RNA was extracted with TRIzol reagent and
reverse transcribed into cDNA. The target genes were then amplified
with the following primers: WWOX (6–8 exons) F,
5′-CACGCATTTTAGAAGAATGG-3′; R, 5′-GACAGCAGCACAGTACACG-3′; GAPDH F,
5′-CAAG GTCATCCATGACAACTTTG-3′; R, 5′-GTCCACCACCC TGTTGCTGTAG-3′.
PCR was performed using a PCR kit (Biomed, Beijing, China), and the
amplifications were carried out in a Mastercycler gradient
thermocycler (Applied Biosystems, USA) as follows: an initial
denaturation for 5 min at 94°C followed by 30 cycles of 94°C for 30
sec, 55°C for 30 sec, 72°C for 30 sec, and a final extension for 7
min at 72°C.
Flow cytometric analysis
The apoptosis ratio (%) was assessed by flow
cytometry (FCM). In brief, infected cells were collected and
stained by Annexin V PE/7-aminoactinomycin D (KeyGEN Biotech,
Nanjing, China) according the manufacturer’s instructions, and
analyzed using a Becton Dickinson FACSCalibur.
Immunofluorescence assay
Concisely, the cell monolayer was fixed with 4%
paraformaldehyde, and incubated at 4°C overnight with rabbit
anti-Wwox (1:500), mouse anti-p53 (1:300) and anti-p73 (1:300),
respectively. FITC-conjugated anti-rabbit and Cy3-conjugated
anti-mouse IgG all diluted at 1:1000. DAPI was used to dye the cell
nuclei. Stained cells were washed by PBS and observed via
fluorescence microscopy.
qPCR analysis
qPCR test was carried out with SYBR Green PCR Master
Mix under the recommended conditions. Primer sequences were
described as: p53 F, 5′-TGCAATAGGTGTG CGTCAGAA-3′; R,
5′-CCCCGGGACAAAGCAAA-3′; p73 F, 5′-AACGCTGCCCCAACCACGAG-3′; R,
5′-GCCGGTT CATGCCCCCTACA-3′; GAPDH F, 5′-GAAGGTGAAGGT CGGAGT-3′; R,
5′-GAAGATGGTGATGGGATTTC-3′. The comparative Ct method was used to
calculate the relative expression level of p73 or p53 as compared
with GAPDH.
Western blot and co-immunoprecipitation
assays
A nuclear and cytoplasmic protein extraction kit was
used for the isolation of proteins from the nucleus and cytoplasm
of the cells. The primary antibodies and their dilutions used were:
rabbit anti-Wwox (1:1000), mouse anti-p53 (1:1000), anti-p73
(1:500), anti-β-actin (1:1000), anti-lamin B (1:1000) and
anti-tubulin (1:1000). Co-immunoprecipitation assay was carried out
according to the protocol of a commercial Co-IP kit. Rabbit
anti-Wwox (2 μg) was added to protein A/G agarose to hook p73 or
p53, and for the mutual detection, 2 μg mouse anti-p73 was used to
hook Wwox or p53.
Statistical analysis
All data are expressed as means ± standard deviation
(SD). Differences between groups were analyzed by the Student’s
t-test or the non-parametric test using SPSS 13.0, and statistical
significance for the data was set at p<0.05.
Results
WWOX was successfully transfected into
Jurkat and K562 cells
We initially examined whether WWOX cDNA was
successfully transfected into Jurkat and K562 cells using
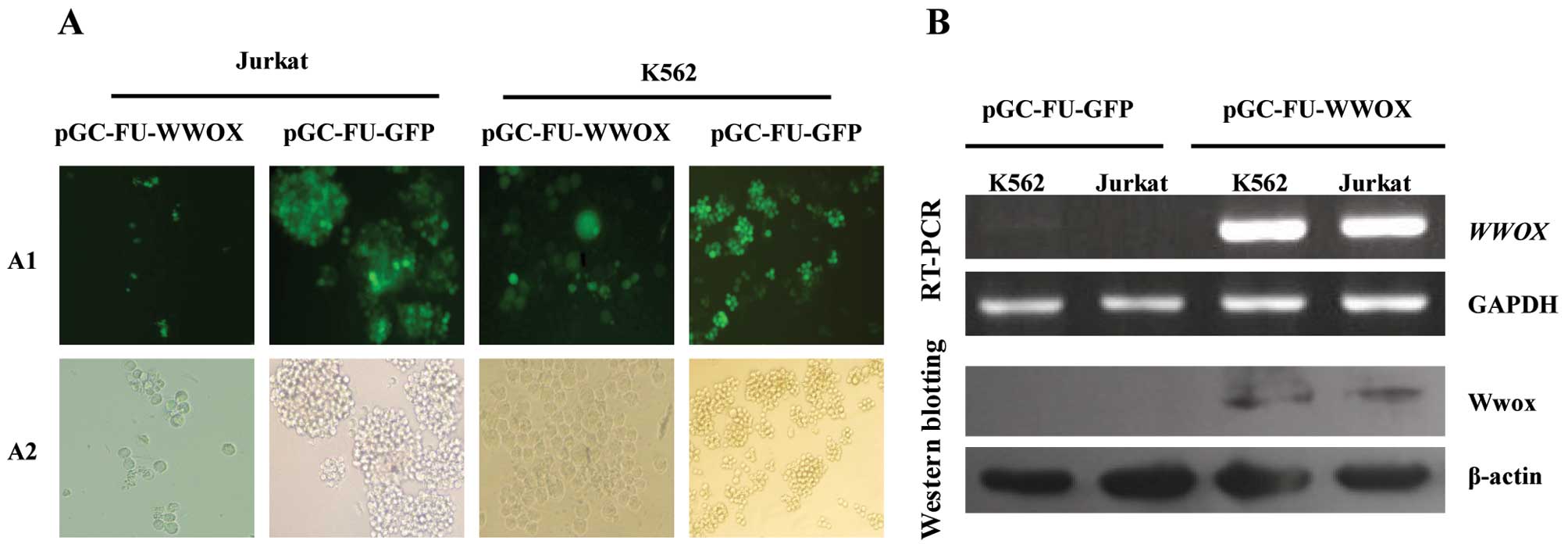
fluorescence microscopy, RT-PCR and western blotting. The results
revealed that cells infected with pGC-FU-WWOX and pGC-FU-GFP were
all observed to express GFP at 48 h after infection (Fig. 1A). RT-PCR and western blot
analysis determined that cells infected with pGC-FU-WWOX exhibited
a high expression level of WWOX mRNA and Wwox protein when
compared with cells transfected with pGC-FU-GFP (Fig. 1B), indicating that WWOX cDNA was
successfully transfected into Jurkat and K562 cells.
WWOX overexpression reduces the viability
of Jurkat and K562 cells
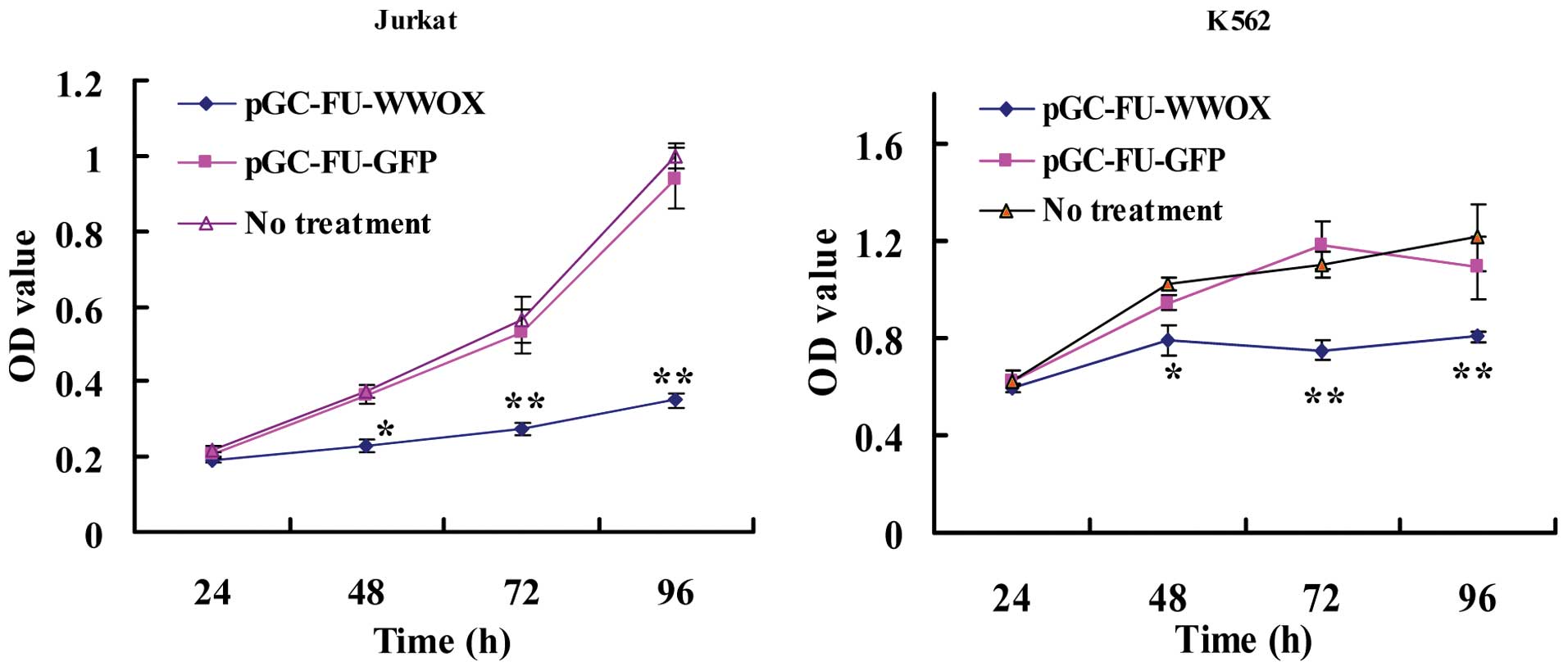
The effects of WWOX overexpression on the viability
of Jurkat and K562 cells was assessed by CCK-8 assay. As shown in
Fig. 2, at 24, 48, 72 and 96 h
following transfection, the OD values (proportional to the cell
numbers) of the pGC-FU-WWOX-infected Jurkat cells were 0.192±0.006,
0.229±0.017, 0.274±0.016 and 0.349±0.020, respectively. Regarding
the K562 cells, the OD values were 0.596±0.017, 0.787±0.06,
0.746±0.039 and 0.803±0.023, respectively; significantly lower when
compared with the OD value of the untreated cells (p<0.05),
while both Jurkat and K562 cells infected with pGC-FU-GFP exhibited
no difference when compared with the untreated cells (p>0.05).
This suggests that WWOX overexpression results in a reduction in
the viability of Jurkat and K562 cells.
WWOX overexpression promotes apoptosis in
Jurkat and K562 cells
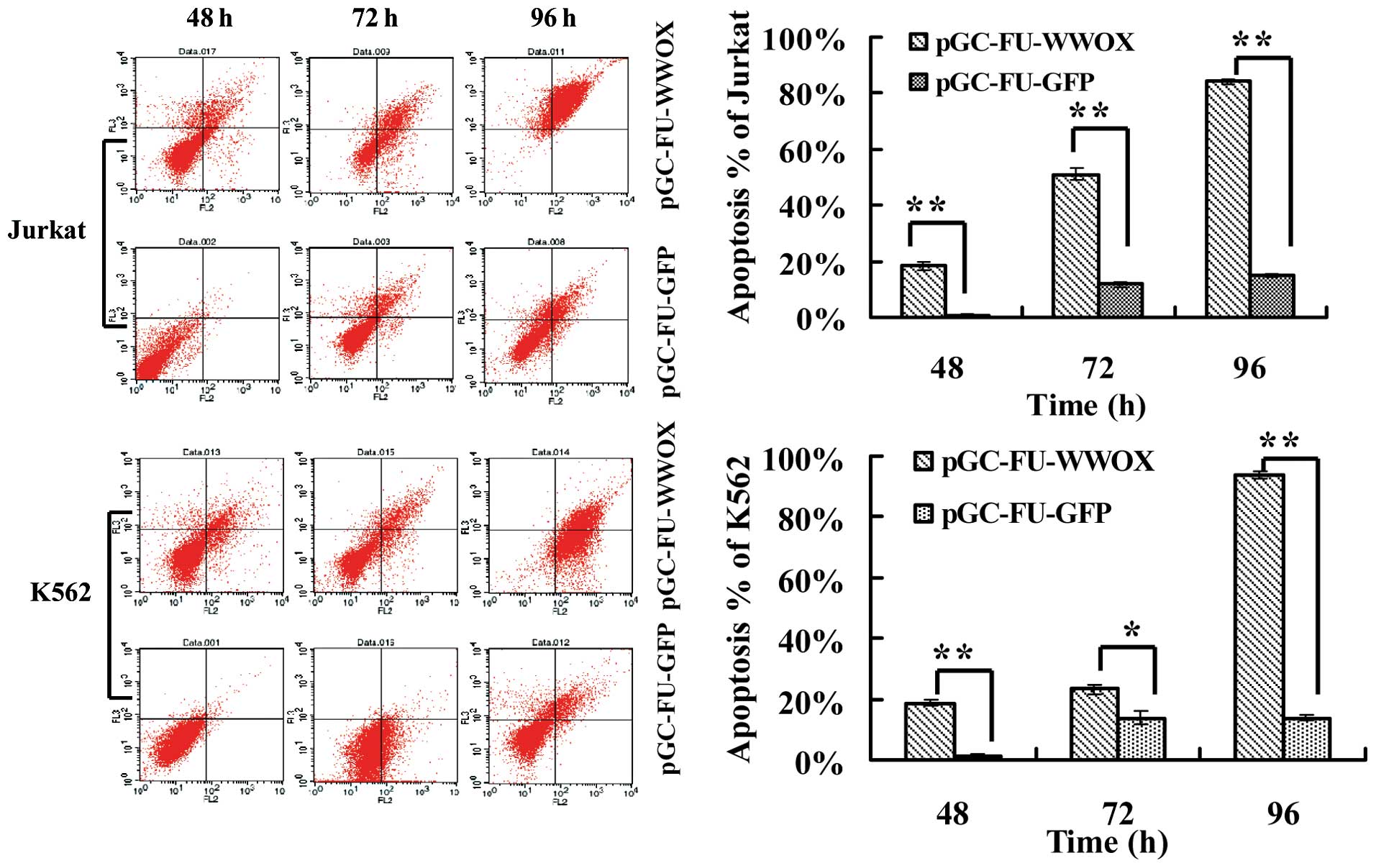
To investigate whether the suppressive effects of
WWOX overexpression on cell viability is due to apoptosis, we
employed FCM after cells were stained with Annexin V
PE/7-aminoactinomycin D. Cells infected with pGC-FU-WWOX exhibited
higher and increased apoptosis ratios (%) when compared with the
apoptosis ratio of cells infected with pGC-FU-GFP during time
lapse. The apoptosis ratio (%) of pGC-FU-WWOX-infected Jurkat cells
was 18.29±1.62% at 48 h, which increased to 84.15±1.10% at 96 h.
Regarding pGC-FU-WWOX-infected K562 cells, the apoptosis ratio was
18.57±1.30% at 48 h, and increased to 93.59±1.26% at 96 h (all with
p<0.05 when compared with the pGC-FU-GFP-infected cells)
(Fig. 3).
WWOX overexpression does not cause a
change in or translocation of p73 and p53
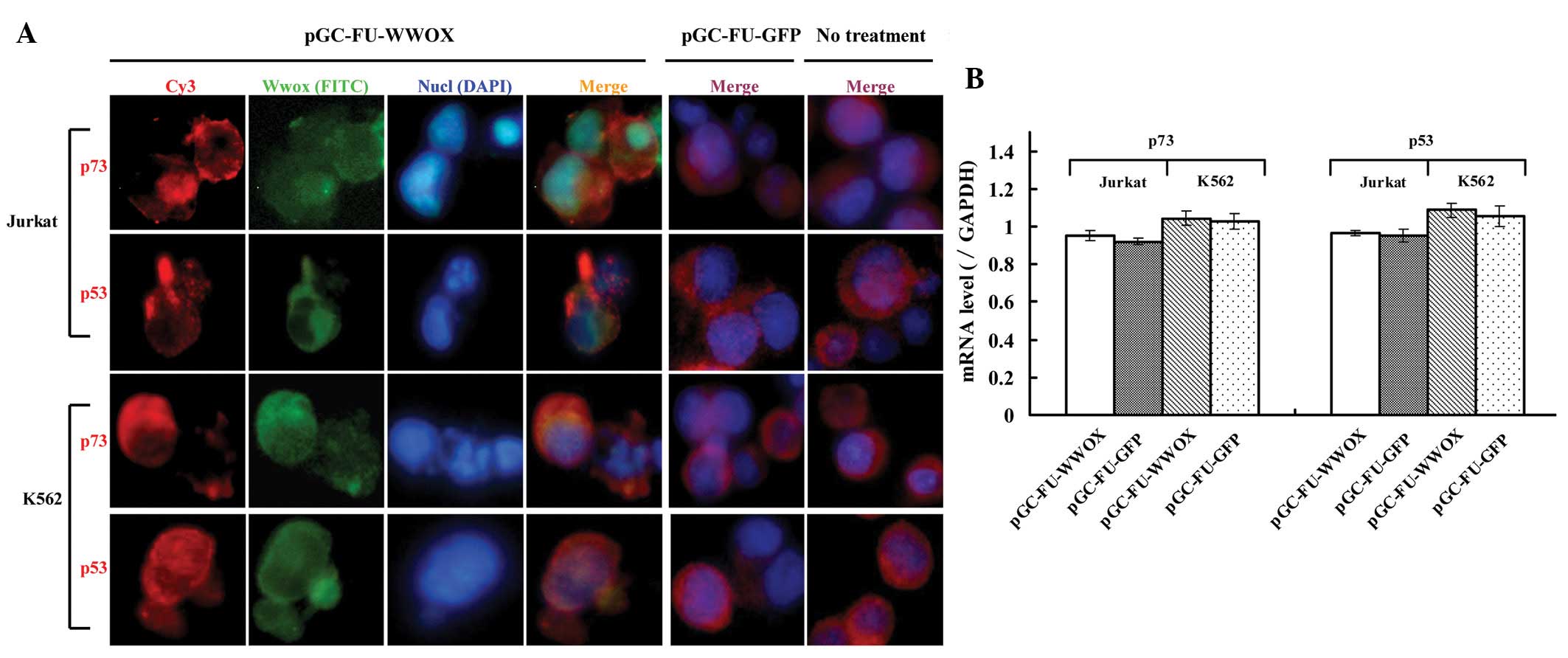
To investigate whether WWOX overexpression causes a
change in expression level or a translocation of p73 or p53 in
Jurkat and K562 cells, we analyzed their subcellular locations via
immunofluorescence assay. Wwox in the pGC-FU-WWOX-infected cells
was located mainly in the cytoplasm, while p73 and p53 were also
mainly located in the cytoplasm. pGC-FU-GFP-infected cells as well
as the untreated cells exhibited similar locations for p73 and p53
when compared with the pGC-FU-WWOX-infected cells (Fig. 4A). We then assessed the changes in
p73 and p53 at the mRNA and protein levels via qPCR and western
blotting. qPCR results revealed that changes in p73 and p53 mRNA in
the pGC-FU-WWOX-infected cells exhibited no difference when
compared with the pGC-FU-GFP-infected cells (p>0.05) (Fig. 4B). Western blot assay demonstrated
that total protein (even in different organelles) of p73 and p53 in
the pGC-FU-WWOX-infected cells was altered inconspicuously compared
with the pGC-FU-GFP-infected cells (p>0.05) (Fig. 4C and D), indicating that WWOX
overexpression was unable to cause a change in translocation of p73
and p53.
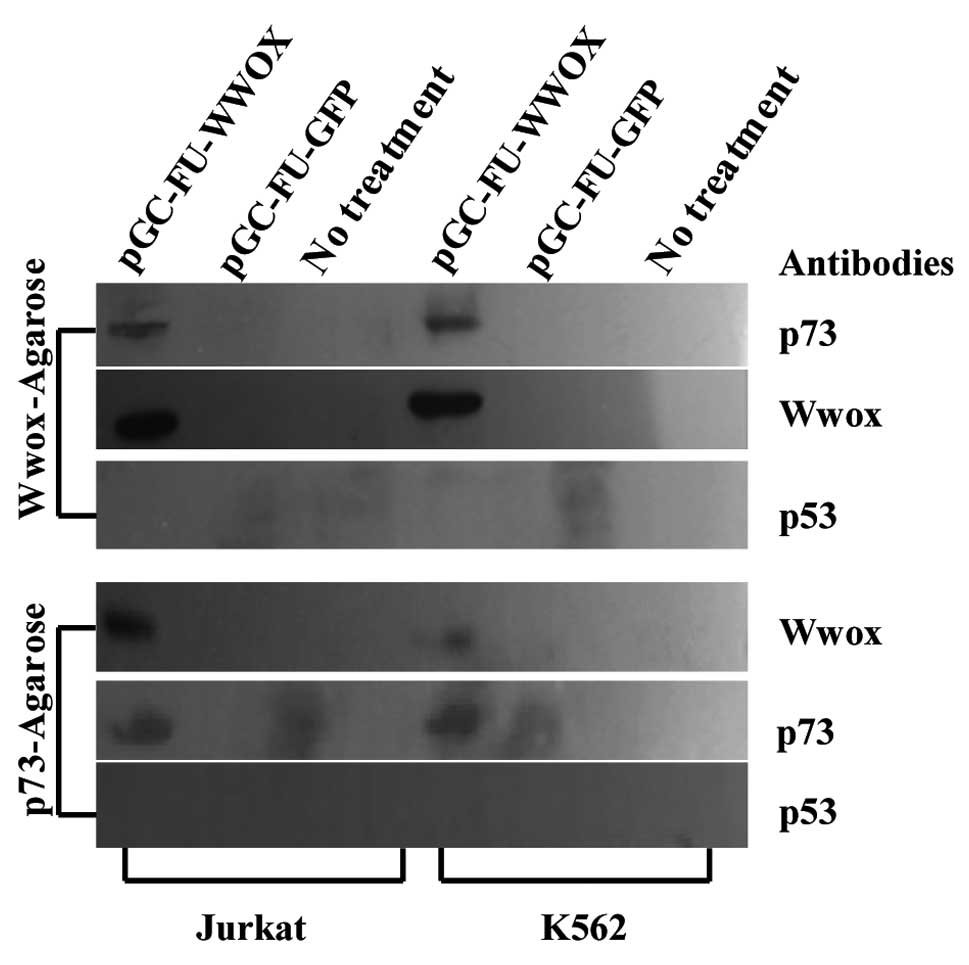
Ectopic Wwox binds with p73 instead of
p53 in the cytoplasm
To investigate the interactions between WWOX and p73
or p53, co-immunoprecipitation and western blot analysis were
applied. Western blot analysis showed that p73 and Wwox in the
precipitation liquid harvested by co-immunoprecipitation
(Wwox-agarose A/G) from pGC-FU-WWOX-infected cells were both
present. At the same time, the mutual detection also showed that
Wwox and p73 in the precipitation liquid harvested by
co-immunoprecipitation (p73-agarose A/G) from the
pGC-FU-WWOX-infected cells were both also detectable. p53 for both
Jurkat and K562 cells was undetectable in this process (Fig. 5).
Discussion
In the present study, we tranfected WWOX cDNA into
human Jurkat and K562 leukemia cell lines using the lentiviral
vector pGC-FU-WWOX, and we explored the effects of WWOX
overexpression on the biological properties of these cell lines.
Our data revealed that WWOX overexpression resulted in significant
supression of cell viability and apoptosis induction in the Jurkat
and K562 cells. We also investigated whether Wwox interacts with
p73 or p53 in regards to its proapoptotic activity. We found that
WWOX binds with p73 instead of p53 in the cytoplasm, indicating
that WWOX has a close relationship with p73 during its proapoptotic
activity in human leukemia.
Reduced WWOX expression and/or aberrant WWOX
mRNA transcripts have been reported in various types of solid
cancers (19–21), and restoration or upregulation of
WWOX in tumor cell lines such as lung, breast and prostate, can
sensitize them to apoptosis (11,12,22), suggesting that WWOX functions as a
tumor-suppressor gene. WWOX also plays an important role in human
hematopoietic malignancies, as aberration or absence of WWOX
expression has been detected in primary hematopoietic malignancies
(23,24). In the present study, we
successfully transfected WWOX cDNA into Jurkat and K562 cells and
observed overexpression of WWOX that resulted in marked inhibition
of cell viability and promotion of apoptosis. Although opposing
views on the function of WWOX as a tumor-suppressor gene exist
(25), the functional concept
that ectopic expression of WWOX leads to apoptosis in leukemia
cells was investigated in our study.
WWOX is reported to have a close relationship with
the tumor-suppressor genes p73 and p53 (14–18,26,27). The first partner of the WW domain
of Wwox to be reported was the p53 homologue named p73, and Wwox
interacts via its first WW domain with the proline-rich motif of
p73 (14). Of note, in a review
by Chang, it was reported that suppression of WWOX expression
abolishes p53-mediated apoptotic function, indicating that WWOX is
a likely partner of p53 in cell apoptosis (26). Gomes et al also found that
WWOX has a close relationship with p53 (18). However, studies by Aqeilan et
al showed that WWOX is able to interact with p73 and suppresses
its transcriptional activity (14,27).
Our data demonstrated that Wwox is able to bind with
cytoplasmic p73 instead of p53, without causing a nuclear-plasma
translocation for p73 in Jurkat and K562 cells. Aqeilan et
al (14) reported that p73
localizes in the nucleus of NIH 3T3 cells, while Wwox localizes to
the cytoplasm, and overexpression of WWOX caused the sequestration
of p73 from the nucleus to the cytoplasm in NIH 3T3 cells; at the
same time, cytoplasmic p73 contributed to the proapoptotic activity
of Wwox.
Another review reported that WWOX and p53 both
co-localize to the cytoplasm, and Wwox binds to the proline-rich
region of p53 via its WW domain (16). Our findings do not agree with the
viewpoints of a previous study (26) and do not agree in part with
another study (14), as we did
not observe a re-localization of p73 from the nucleus to the
cytoplasm or from the cytoplasm to the nucleus in the
pGC-FU-WWOX-infected cells, as well as interactions between Wwox
and p53. Yet, our findings are supported in part and fully by
previous studies (14,27).
One of the possible explanations is that the
translocation of p73 or p53 in different types of cell lines is
inherently different. For instance, p73 mainly localizes in the
cytoplasm of Jurkat and K562 cells, and the nucleus exhibits p53 to
a small extent, while in NIH 3T3 cells, p73 exhibits a high
expression level in the nucleus (14). However, in our study we observed
that Wwox and p73 co-localize in the cytoplasm of both Jurkat and
K562 cells and bind together. Although p53 was not observed to bind
with Wwox in our experiment, it is not proved that they have no
interactions, and may probably exist in another manner. Notably, we
found a high expression level of p53 in both cell lines although
they were between passages 15 and 30. The most likely explanation
is that the p53 protein was not encoded by the wild-types, but the
mutant ones, which indicated that there might exist mutations of
p53 in Jurkat and K562 cells. Nevertheless, it is unclear whether
the p53 protein in our samples was wild-type or not, and more
evidence is needed.
In summary, this is the first study to upregulate
Wwox expression in human leukemia, in order to uncover the
preliminary mechanisms of WWOX-mediated apoptosis in Jurkat and
K562 cells. We demonstrated that WWOX has a close relationship with
p73 in its proapoptotic activity in leukemia. However, our study
did not trace the actual binding sites between WWOX and p73, as
well as the phosphorylated levels for p73 and p53 expression.
Furthermore, a mammalian two hybrid assay or GST-pull down analysis
is needed. These experiments will be performed in future studies by
our group.
Acknowledgements
This study was supported by the Provincial Natural
Science Fund of Fujian, grant no. 2010J01181.
References
|
1
|
Bednarek AK, Laflin KJ, Daniel RL, Liao Q,
Hawkins KA and Aldaz CM: WWOX, a novel WW domain-containing protein
mapping to human chromosome 16q23.3–24.1, a region frequently
affected in breast cancer. Cancer Res. 60:2140–2145.
2000.PubMed/NCBI
|
|
2
|
Ried K, Finnis M, Hobson L, et al: Common
chromosomal fragile site FRA16D sequence: identification of the FOR
gene spanning FRA16D and homozygous deletions and translocation
breakpoints in cancer cells. Hum Mol Genet. 9:1651–1663. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Del Mare S, Salah Z and Aqeilan RI: WWOX:
its genomics, partners, and functions. J Cell Biochem. 108:737–745.
2009.PubMed/NCBI
|
|
4
|
Macias MJ, Wiesner S and Sudol M: WW and
SH3 domains, two different scaffolds to recognize proline-rich
ligands. FEBS Lett. 513:30–37. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Nunez MI, Ludes-Meyers J and Aldaz CM:
WWOX protein expression in normal human tissues. J Mol Hist.
37:115–125. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang X, Chao L, Jin G, Ma G, Zang Y and
Sun J: Association between CpG island methylation of the WWOX gene
and its expression in breast cancers. Tumour Biol. 30:8–14. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Guo W, Wang G, Dong Y, Guo Y, Kuang G and
Dong Z: Decreased expression of WWOX in the development of
esophageal squamous cell carcinoma. Mol Carcinog. Dec 27–2011.(Epub
ahead of print).
|
|
8
|
Nakayama S, Semba S, Maeda N, Matsushita
M, Kuroda Y and Yokozaki H: Hypermethylation-mediated reduction of
WWOX expression in intraductal papillary mucinous neoplasms of the
pancreas. Br J Cancer. 100:1438–1443. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dias EP, Pimenta FJ, Sarquis MS, et al:
Association between decreased WWOX protein expression and thyroid
cancer development. Thyroid. 17:1055–1059. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yang J, Cogdell D, Yang D, et al: Deletion
of the WWOX gene and frequent loss of its protein expression in
human osteosarcoma. Cancer Lett. 291:31–38. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Fabbri M, Iliopoulos D, Trapasso F, et al:
WWOX gene restoration prevents lung cancer growth in vitro and in
vivo. Proc Natl Acad Sci USA. 102:15611–15616. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Iliopoulos D, Fabbri M, Druck T, Qin HR,
Han SY and Huebner K: Inhibition of breast cancer cell growth in
vitro and in vivo: effect of restoration of Wwox expression. Clin
Cancer Res. 13:268–274. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Aqeilan RI, Trapasso F, Hussain S, et al:
Targeted deletion of Wwox reveals a tumor-suppressor function. Proc
Natl Acad Sci USA. 104:3949–3954. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Aqeilan RI, Pekarsky Y, Herrero JJ, et al:
Functional association between Wwox tumor suppressor protein and
p73, a p53 homolog. Proc Natl Acad Sci USA. 101:4401–4406. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hezova R, Ehrmann J and Kolar Z: WWOX, a
new potential tumor suppressor gene. Biomed Pap Med Fac Univ
Palacky Olomouc Czech Repub. 151:11–15. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yang JL and Zhang W: WWOX tumor suppressor
gene. Histol Histopathol. 23:877–882. 2008.
|
|
17
|
Chang NS, Hsu LJ, Lin YS, Lai FJ and Sheu
HM: WW domain-containing oxidoreductase: a candidate tumor
suppressor. Trends Mol Med. 13:12–22. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gomes CC, Diniz MG, Oliveira CS, Tavassoli
M, Odell EW, Gomez RS and De Marco L: Impact of WWOX alterations on
p73, ΔNp73, p53, cell proliferation and DNA ploidy in salivary
gland neoplasms. Oral Dis. 17:564–571. 2011.
|
|
19
|
Aqeilan RI, Kuroki T, Pekarsky Y, et al:
Loss of WWOX expression in gastric carcinoma. Clin Cancer Res.
10:3053–3058. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Alsop AE, Taylor K, Zhang J, Gabra H,
Paige AJ and Edwards PA: Homozygous deletions may be markers of
nearby heterozygous mutations: the complex deletion at FRA16D in
the HCT116 colon cancer cell line removes exons of WWOX. Genes
Chromosomes Cancer. 47:437–447. 2008. View Article : Google Scholar
|
|
21
|
Żelazowski MJ, Płuciennik E, Pasz-Walczak
G, Potemski P, Kordek R and Bednarek AK: WWOX expression in
colorectal cancer: a real-time quantitative RT-PCR study. Tumour
Biol. 32:551–560. 2011.PubMed/NCBI
|
|
22
|
Qin HR, Iliopoulos D, Semba S, et al: A
role for the WWOX gene in prostate cancer. Cancer Res.
66:6477–6480. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ishii H and Furukawa Y: Alterations of
common chromosome fragile sites in hematopoietic malignancies. Int
J Hematol. 79:238–242. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ishii H, Vecchione A, Furukawa Y, et al:
Expression of FRA16D/WWOX and FRA3B/FHIT genes in hematopoietic
malignancies. Mol Cancer Res. 1:940–947. 2003.PubMed/NCBI
|
|
25
|
Watanabe A, Hippo Y, Taniguchi H, et al:
An opposing view on WWOX protein function as a tumor suppressor.
Cancer Res. 63:8629–8633. 2003.PubMed/NCBI
|
|
26
|
Chang NS: A potential role of p53 and WOX1
in mitochondrial apoptosis (Review). Int J Mol Med. 9:19–24.
2002.PubMed/NCBI
|
|
27
|
Aqeilan RI, Donati V, Palamarchuk A, et
al: WW domain-containing proteins, WWOX and YAP, compete for
interaction with ErbB-4 and modulate its transcriptional function.
Cancer Res. 65:6764–6772. 2005. View Article : Google Scholar : PubMed/NCBI
|