Introduction
Osteoarthritis (OA), a highly prevalent, slowly
progressive, degenerative disease of diarthrodial joints, is
characterized by a progressive degradation of articular cartilage
associated with marginal osteophyte formation, progressive
symptomatic loss of mechanical function and remodeling of the
subchondral bone, belonging to the GU BI of Traditional Chinese
Medicine (TCM) (1,2). As the precise molecular mechanism of
OA has yet to be fully elucidated, a wide variety of animal models
have been developed to study osteoarthritic progression,
characterize the features of the early pathological changes of OA
and to evaluate new drugs and/or original therapies (3). The papain-induced OA model has been
widely studied in various animal species such as rats, thus
providing new insights into pathogenic mechanisms and impact of
hyaline cartilage (4,5). In this model, there is a sequence of
events in which the degradation of the superficial zone develops
into fibrillations of the cartilage and eventually leads to
ulcerations, erosions and tidemark replication.
Tidemark, at which non-mineralized cartilage comes
to contain hydroxyapatite, is a chondro-osseous junction between
cartilage and bone in diarthrodial joints (6). The vicinity of the tidemark enhanced
metabolic activity, consistent with mineralization, including the
expression of alkaline phosphatase, the deposition of type X
collagen and the ability to bind tetracycline in vivo. The
tidemark replication is considered a characteristic of the
osteoarthrotic process with the advance of a calcification front
advancing into the non-calcified cartilage of zone IV (7). The changes of tidemark are
considered to be coordinated with the resorption of the calcified
cartilage of zone V, as the subchondral bone thickens and replaces
it (8).
The loss of chondrocyte function was found to be a
persistent and important event in OA. A variety of stimuli, such as
mechanical injury, loss of growth factors or excessive reactive
oxygen species, can induce chondrocyte depletion (9). Since chondrocyte is solely
responsible for the maintenance and production of extracellular
matrix (ECM), chondrocyte depletion is indicated in the cartilage
degradation, which pertains to OA pathogenesis (10,11). Chondrocyte apoptosis was thought
to be a major cause of chondrocyte depletion during OA progression,
so enhanced chondrocyte apoptosis is considered to be a sign of
progressive cartilage degradation. However, the extent of the
contribution of apoptotic cell death to chondrocyte depletion in OA
progression remains to be resolved. Previous studies reported that
another type of cell death, autophagy, may be involved in
chondrocyte death during OA progression (12,13).
Tougu Xiaotong capsule (TXC), a TCM formulation,
consists of a combination of four natural products including
Radix Morindae Officinalis, Radix Paeoniae Alba,
Rhizoma Chuanxiong and Glabrous Sarcandra Herb. These natural
products together confer TXC properties of nourishing Shen,
supplementing Jing, filling in Sui, stretching tenders and dredging
collaterals to strengthen tendons and bones at the theories of TCM.
TXC has been used for the osteoarthritic treatment in the Second
People's Hospital Affiliated to Fujian University of TCM for two
decades, and it has been shown to have significant therapeutic
effects on OA in the clinical trials, such as evident improvements
in osteoarthritic symptoms, pain, swelling and motion of joint.
Previously, we reported that TXC could inhibit chondrocyte
apoptosis by upregulation of Bcl-2, downregulation of p53,
caspase-9 and caspase-3 (14).
However, the molecular mechanism of the therapeutic effect of TXC
remains largely unknown. Therefore, using a papain-induced OA in
rat knee joints, we evaluated the effect of TXC on the tidemark
replication and cartilage degradation, and investigated the
underlying mechanisms of TXC in the regulation of chondrocyte
autophagy.
Materials and methods
Animals
Forty 4-week-old male Sprague-Dawley (SD) rats of
Specific Pathogen Free (SPF), qualified number SCXK (Shanghai)
2007-0005, were purchased from the Shanghai Slack Laboratory Animal
Co. (Shanghai, China). The Fujian University of TCM Experimental
Animal Centre offers SPF medical laboratory animal environmental
facilities, qualified number SYXK(Min) 2009-0001. The care and use
of the laboratory animals complied with the Guidance Suggestions
for the Care and Use of Laboratory Animals 2006 of the Ministry of
Science and Technology, China.
Experimental design
After one week of acclimation, the rats received a
12 μl intra-articular injection of a 1 U/ml of L-cysteine-activated
papain in phosphate buffered saline (PBS) (Sigma, St. Louis, MO,
USA) in their double knee joints at 1, 4 and 7 days (4). Eight weeks after papain-induced OA,
the animals were randomly divided into four groups. The TXC+OA
group received oral TXC (the Second People's Hospital Affiliated to
Fujian University of TCM, medical license no. MINZHIZI Z20100006;
184 mg/kg/day). The glucosamine (GlcN)+OA group received oral GlcN
sulfate (Sigma; 150 mg/kg/day). The OA group and control group (non
papain-induced OA) received equivalent saline only. All groups were
treated once a day for 12 consecutive weeks, following which the
animals were sacrificed. The changes of cartilage structure and
tidemark were observed by digital radiography (DR), optical
microscopy, scanning electron microscopy (SEM) and transmission
electron microscopy (TEM).
Gross morphology of the knee joints
The gross morphological changes in cartilage were
examined by DR (LDRD-01BL, Beijing Aerospace Zhongxing Medical
System Co., Ltd., China), and the grade of knee joint degradation
of DR films was according to the Kellgren-Lawrence X-ray grade
standard (15).
The tibial plateau cartilage was bivalved in the
coronal plane with a sharp osteotome. The exposed surface was
rinsed with PBS repeatedly to remove blood and bone marrow.
Specimens, 5 mm × 5 mm × 5 mm in size for SEM, were then fixed with
2% glutaraldehyde solution, washed with 0.1 M sodium cacodylate
buffer, and post-fixed with 1% osmium tetroxide. After dehydrating
with an alcohol gradient series, and dehydrating with isoamyl
acetate again, the specimen was dried using a critical point dryer
with HCP-2. After coating with a layer of gold, all specimens were
observed under SEM (JSM-6380LV, JEOL, Japan).
Histopathological examination of the knee
joints
The joints were sectioned 0.5 cm above and below the
joint line, fixed in 10% neutral buffered formalin for 3 days, and
then decalcified for 2 weeks in buffered 12.5%
ethylenediaminetetraacetic acid (EDTA) and formalin solution. The
cartilage was stained with Safranin O-fast green stains to assess
the general morphology and matrix proteoglycans. Cartilage
histological changes were evaluated according to the Mankin score
(16).
Ultrastructural examination of the knee
articular cartilage
The tibial plateau cartilage, 2 mm × 2 mm × 2 mm in
size for TEM, was fixed in 3% glutaraldehyde and 1.5%
paraformaldehyde solution (pH 7.3) at 4°C for 24 h, postfixed with
1% osmic acid and 1.5% potassium hexacyanoferrate (II) solution (pH
7.3) at 4°C for 2 h. The samples were then washed, dehydrated with
graded alcohol, and embedded in Epon-Araldite resin. Ultrathin
sections were cut on a Leica ultramicrotome and stained with 2%
aqueous uranyl acetate, counter stained with 0.3% lead citrate and
examined with TEM (H7650, JEOL).
The subchondral bone was dried at 180°C for 24 h to
grind and pass the powder through 200-mesh steel sieve, and
examined with High Resolution TEM (JEM-2010, JEOL).
Western blot analysis
Protein (20 μg) of each sample was heated to 100°C
for 5 min and then resolved on a 10% sodium dodecyl sulfate
polyacrylamide gel electrophoresis (SDS-PAGE) gel. The proteins
were transferred to methanol-wetted polyvinylidene difluoride
(PVDF) membranes in Tris/Glycine transfer buffer. Subsequently, the
membranes were blocked for 1 h at room temperature in blocking
buffer (5% skim milk powder, 0.5% Tween-20 in tris-buffered saline;
TBS). Blots were incubated with LC3 I/II, ULK1, Beclin1 and β-actin
(Abcam, Cambridge, UK) followed by an HRP-conjugated secondary
antibody. Immunoreactive proteins were visualized by Western Blot
Chemiluminescence Luminol Reagent (Santa Cruz Biotechnology, Santa
Cruz, CA, USA). Immunoblot bands were quantitated with the Tocan
190 protein assay system (Bio-Rad, USA).
Statistical analysis
All data are represented as the means of averages ±
standard deviation (SD) and analyzed by using the SPSS package for
Windows (version 13.0). Statistical analysis of the data was
performed with Student's t-test and ANOVA. The enumeration data was
analyzed by the Chi-square test. P<0.05 was considered to
indicate statistically significant differences.
Results
No signs of drug toxicity were noted in the SD rats
treated with TXC or GlcN. The level of daily activity was similar
in all four experimental groups, and there were no significant
differences in body weight between the groups during the study
period.
TXC delays the degradation of
papain-induced OA
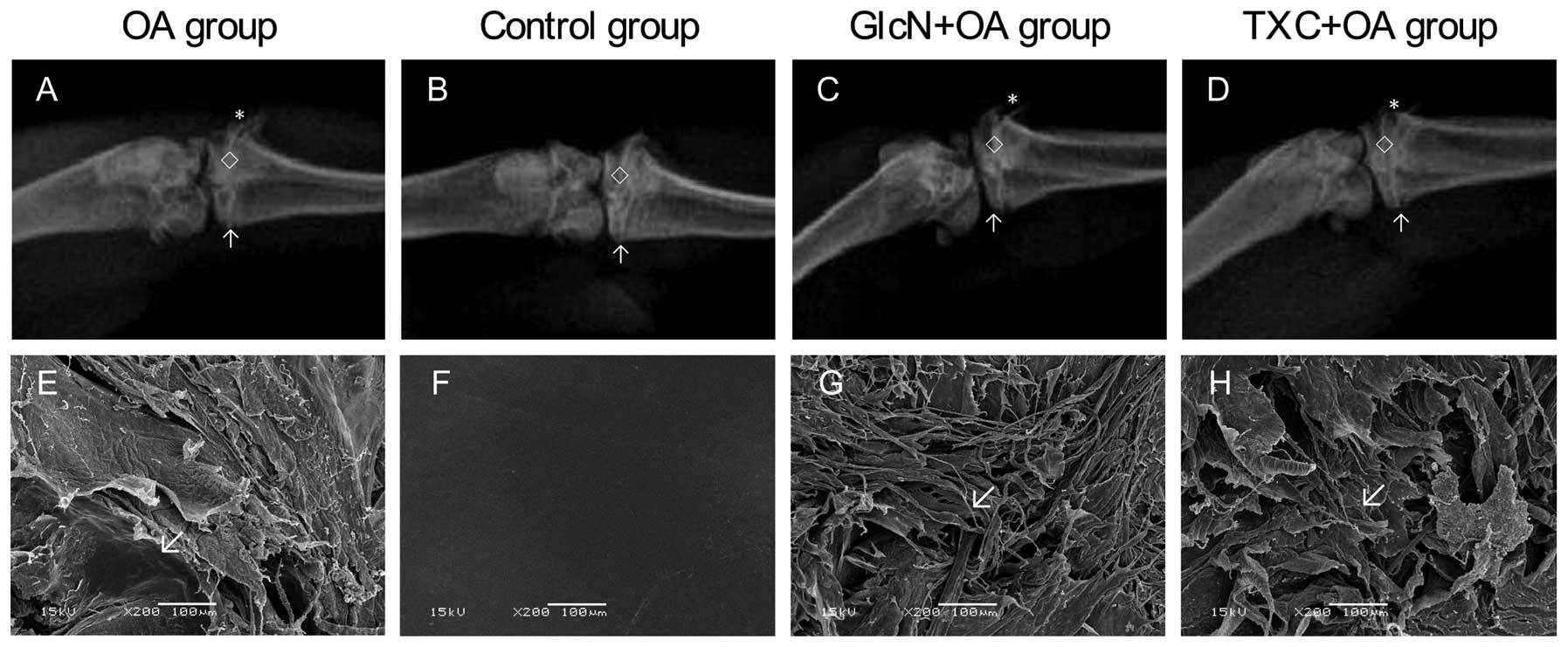
In the knee joints, gross morphologic changes with a
significant difference between the control group and the treatment
with TXC or GlcN and OA groups are shown in Fig. 1. The width of the hind limb knee
joint of the TXC or GlcN group was clearer and wider than that of
the OA group. In the OA group, gross morphologic changes were
characterized by cartilage degradation, such as fibrillation,
erosion and ulcer formation, and osteophyte formation, observed in
the femoral condyle and tibial plateau. Markedly less severity of
knee joint degradation was observed following treatment with TXC or
GlcN (Table I; P<0.05).
 | Table IKellgren-Lawrence X-ray grade standard
in the different groups. |
Table I
Kellgren-Lawrence X-ray grade standard
in the different groups.
| Group | G0 | G1 | G2 | G3 | G4 |
|---|
| OA | 0 | 0 | 0 | 4 | 6 |
| Controlb | 10 | 0 | 0 | 0 | 0 |
|
GlcN+OAa,c | 0 | 0 | 2 | 6 | 2 |
| TXC+OAa,c | 0 | 0 | 3 | 5 | 2 |
TXC inhibits cartilage tidemark
replication
Tibial plateau cartilage from the OA group showed
evident histological changes, such as moderate-to-severe
hypocellularity, complete disorganization, proteoglycan reduction
on Safranin O-fast green staining of ECM, denudation of articular
surface and fissures extending into the deep zones, and tidemark
replication, compared to the control group (Fig. 2A–F). Osteophytes were present at
the medial margins of the tibial plateau. In turn, tibial plateau
cartilage from the control group showed homogeneous and intense
staining of proteoglycans at ECM, normal cellularity and structure
across the different layers, and tidemark wave-shaped (Fig. 2G and H). As shown in Fig. 2I–L, tibial plateau cartilage
following the treatment with TXC or GlcN displayed improvement in
cellularity and cellular organization in chondron-like manner,
although there was some diffuse hypercellurarity compared to the OA
group. There was also reduction of structure irregularities and
mild improvement of ECM. The Mankin scores of cartilage in both the
TXC or GlcN and OA groups were significantly higher than those in
the control group (P<0.01), and in the TXC or GlcN groups they
were significantly lower than those in the OA group (P<0.01)
(Fig. 2M).
 | Figure 2Histology analyses of the medial
tibial plateau treated with TXC. Histological sections of the
tibial plateau stained with Safranin O-fast green to illustrate
pathological changes. The OA tibial plateau showed loss of viable
chondrocytes (A, arrow), chondrocyte proliferation (B, asterisk),
loss of proteoglycans, and tidemark replication (C and D, arrow).
The cartilage showed fibrillation, vertical fissures (E, diamond)
and delamination (F). The subchondral trabecular bone architecture
was altered with sclerosed bone (C and D, asterisk) and the
cellular bone marrow was replaced by loosely arranged spindle cells
in a fine fibrous stroma (B and E, arrow). The control tibial
plateau showed normal healthy cartilage with normally distributed
chondrocytes (G) and tidemark wave-shaped (H, arrow). The red
coloration of the hyaline cartilage, reflecting proteoglycan
content, is homogenous and the surface of the cartilage is smooth.
The GlcN+OA tibial plateau showed the cartilage is lost, the
hyaline cartilage has reduced proteoglycans (loss of staining), a
reduced thickness (erosion) is diffusely hypocellular and clones
are present at the periphery of the changes (I and J). There
appears to be an increase in the number and size of bone lacunae
under the pits. The overlying hyaline cartilage in TXC+OA has
increased proteoglycans compared to the OA cartilage (K and L).
Mankin score analysis of the medial tibial plateau (M). Symbols
represent the means of averages ± SD and SD is shown as vertical
bars. **P<0.01, significant difference vs. the OA
group; ☆☆P<0.01, significant difference vs. the
control group. |
TXC delays chondrocyte and subchondral
bone degradation
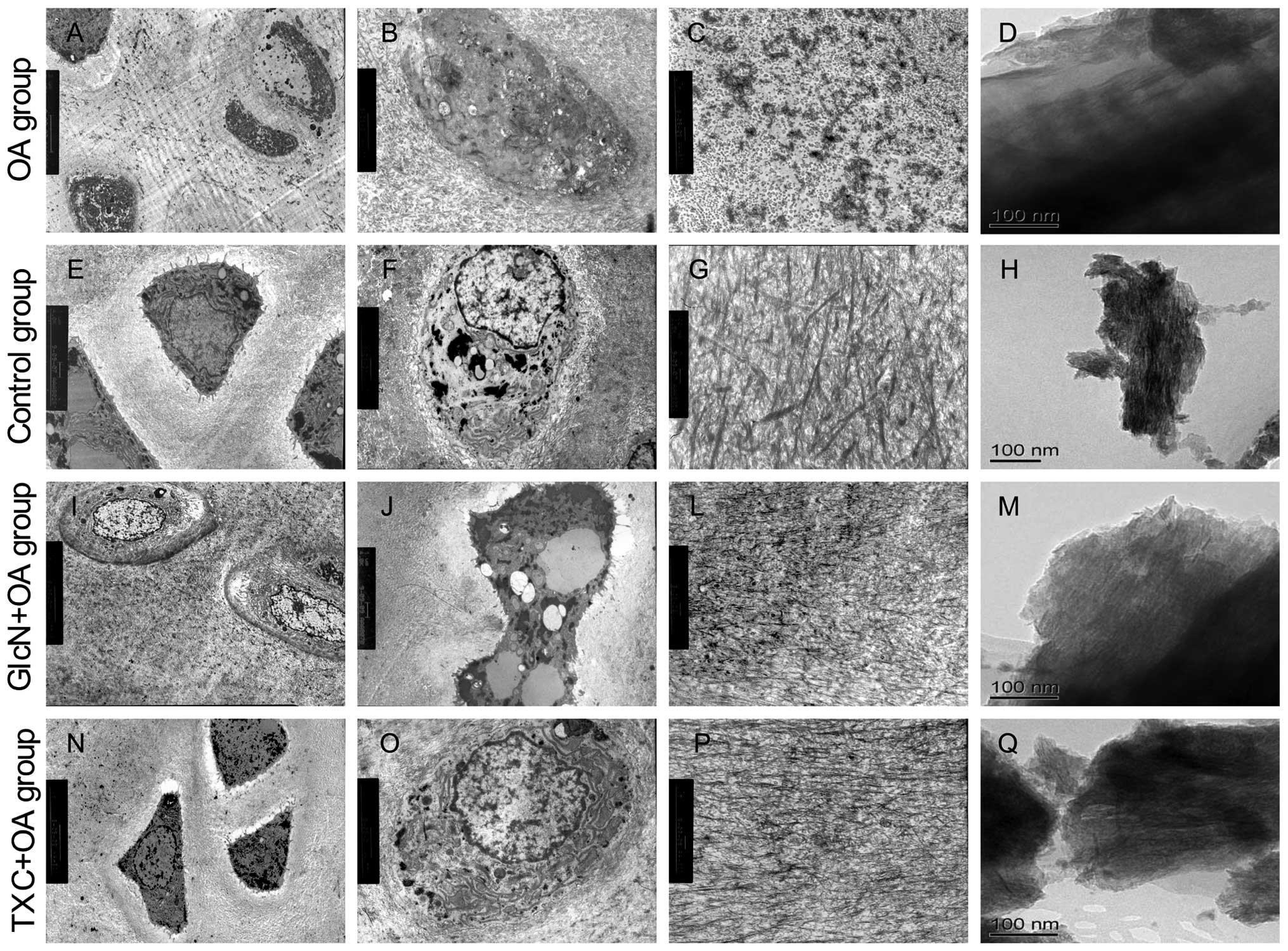
Ultrastructural study of tibial plateau cartilage in
both the TXC or GlcN and OA groups by TEM revealed distinctive
differences compared to control cartilages. The cartilage in the OA
group showed microscopic evidence of surface fibrillation, loss of
ECM staining in the superficial region for proteoglycans, several
apoptotic cells and reduced cellularity. Apoptotic cells were
shrunken and clearly retracted from the surrounding ECM. The
ultrastructural characteristics of apoptotic cells that we observed
were the presence of nuclear blebbing, apoptotic bodies and cell
shrinkage, whereas intensified staining of the cytoplasm, blebbing
of the cell membrane, and condensation of the chromatin were
observed less frequently (Fig. 3A and
B). The ECM contained degradation of several collagen fibers
(Fig. 3C).
The subchondral bone showed loosening and
irregularity of collagen arrangement, dense crystals of calcium and
phosphorus (Fig. 3D). The
cartilage in the control group showed chondrocytes were generally
round and characterized by several microvilli-like structures and
corrugations at the chondrocytic surface, some cells with the
characteristics of autophagosomes (Fig. 3E). The cytoplasm contained some
mitochondria and a relatively abundant number of organelles assumed
either rod-like or spherical morphology, and presented clearly
visible cristae. The nucleus of chondrocytes showed lobate or
indentation morphology, heterochromatin was observed preferentially
at the periphery of the nucleus (Fig.
3F). The ECM typically consists of a network of tightly packed
and highly cross-linked collagen fibrils (Fig. 3G). The subchondral bone showed
regularity of collagen arrangement, uniform distribution of calcium
phosphate crystals (Fig. 3H).
Markedly less severity of cartilage and subchondral bone
degradation was observed in the treatment with TXC or GlcN
(Fig. 3I–Q).
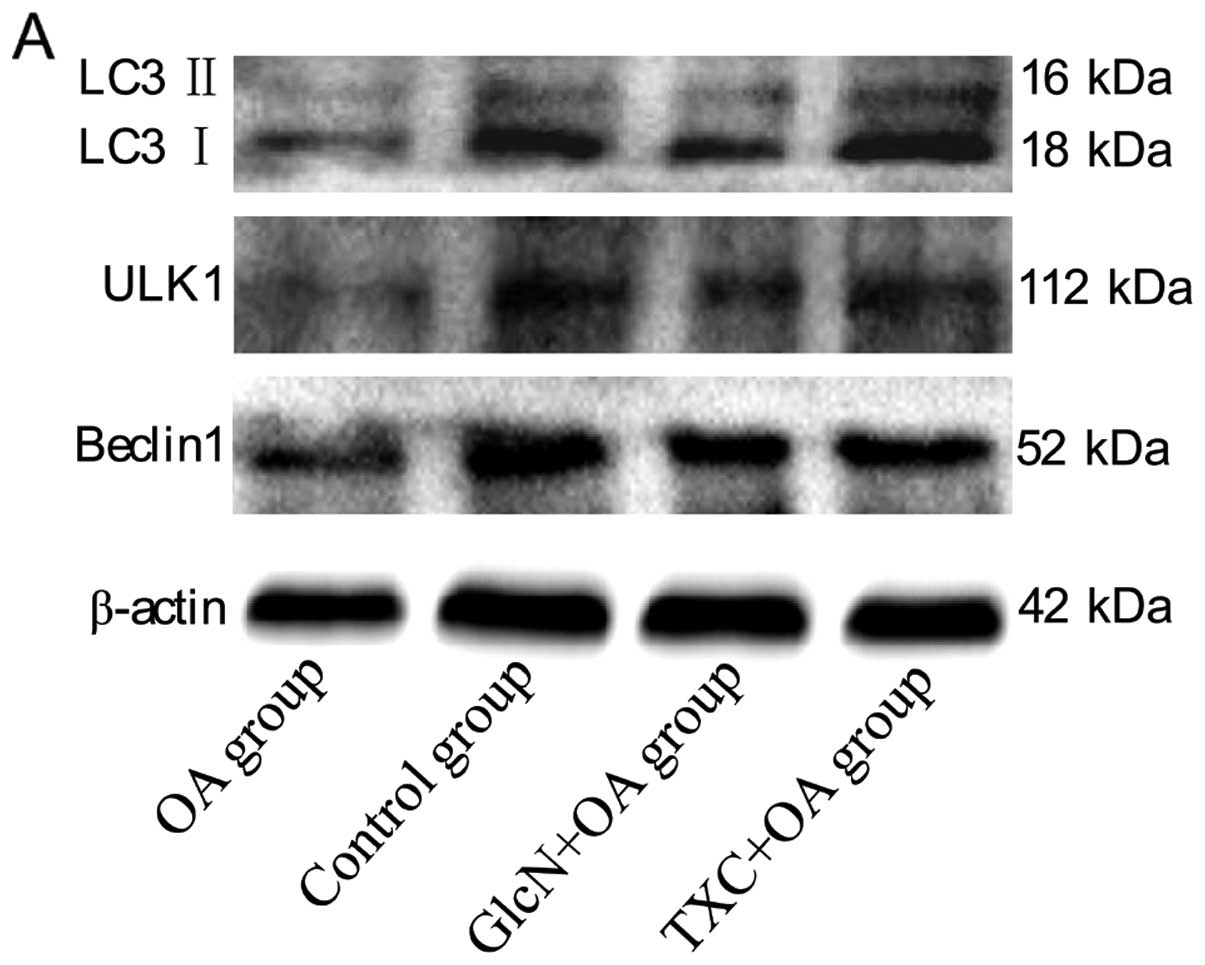
TXC enhances the autophagy-related
proteins against chondrocyte apoptosis
We then examined whether another type of cell death,
autophagy, was involved in OA pathogenesis. We first examined the
conversion of LC3 from an 18 kDa form (LC3 I) to a faster-migrating
16 kDa form (LC3 II), ULK1 and Beclin1. As shown in Fig. 4A–E, a western blot assay
demonstrated a decrease in LC3 I/II, ULK1 and Beclin1 levels in the
OA group compared to those in the control group (P<0.01).
However, LC3 I/II, ULK1 and Beclin1 levels in both the TXC and the
GlcN group significantly increased compared to those in the OA
group (P<0.05, P<0.01).
Discussion
The present study systematically evaluated the
cartilage protection mechanisms of TXC in papain-induced OA. Our
results clearly showed that TXC could improve the arrangement of
subchondral bone collagen fibers and calcium phosphate crystals,
inhibit the tidemark replication and delay the cartilage
degradation. In addition, our study showed that TXC upregulated the
protein levels of LC3 I/II, ULK1 and Beclin1, demonstrating that it
could inhibit the tidemark replication and cartilage degradation by
the activation of chondrocyte autophagy.
A number of treatment programs of OA have been
developed, such as medications with NSAIDs and chondroprotective
drugs. However, major problems associated with medications still
remain, particularly with the side-effects of NSAIDs. Thus, there
is a pressing need to develop alternative approaches to OA
treatment (17). Complementary
and alternative medicine is currently of interest to the general
public and the medical profession, and includes TCM. Chinese herbs
have relatively fewer-side effects compared to modern
chemotherapeutics and have long been used clinically to treat OA.
TXC, a famous TCM formulation, has been reported to be clinically
effective in treating OA by inhibiting chondrocyte apoptosis
(14). However, the molecular
mechanism of the therapeutic effect of TXC remains largely unknown.
Therefore, the present study examined whether TXC regulates the
tidemark replication and cartilage degradation by the regulation of
chondrocyte autophagy.
Tidemark, a distinct boundary between non-calcified
and calcified articular cartilage and not an artifact, has been
described as a haematoxyphil single line which is approximately
10-μm thick (18,19). Chondrocytes near the tidemark must
regulate the turnover of non-collagenous components in ECM and
maintain control over the local ECM (20). During normal development and
growth of diarthrodial joints, the tidemark clearly represents a
calcification front. In the normal adult joint, the tidemark is
still a single structure, ceases the advance of mineral into the
hyaline cartilage, although a residual ‘maintenance’ turnover of
ECM may occur (21). Under these
conditions, although the tidemark still contains some tightly bound
calcium, it may have ceased to function as a calcification front.
It is possible that the tidemark has changed in function to one of
inhibiting the growth or formation of microcrystals of
hydroxyapatite result in protecting the hyaline cartilage from
passive progressive mineralization at this stage. This may be an
irreversible change, so that if new mineralization is activated in
OA, a new tidemark may actively form distally to the original one,
leaving its predecessor as a non-functional relic, and thus
providing an explanation of tidemark replication. The tidemark
replication in OA is characterized by an endochondral ossification
process advancing calcification of zone IV and replacement of
calcified cartilage by new bone at the calcified cartilage-bone
interface, and these events must be precisely determined and
regulated by adjacent chondrocytes (22). Histological measures of articular
cartilage pathology, generally considered to be the reference
standard for presence and severity of OA, showed significant
tidemark replication and cartilage degradation in the OA group
compared to the control group, indicating that OA was successfully
induced by papain. In the present study, the TXC+OA group showed a
smaller increase in knee joint width and an inhibition of cartilage
degradation as compared to the OA group. Collectively, the above
findings support a role for TXC in the protection of cartilage and
chondrocyte metabolism, suggesting a possible mechanism by which
TXC may help to alleviate clinical signs and retard progression of
OA.
OA is a characterized by cartilage degradation and
subchondral bone changes. Microcracks in the calcified tissues may
enhance cellular activity leading to increased bone remodeling
(23,24). Although the relationship between
cartilage degradation and subchondral bone changes remains
controversial, the subchondral bone is considered to play an
important role in OA initiation and progression. During the OA
process, the subchondral bone would then become stiffer, causing a
reduced shock-absorbing capacity and leading to progression of
these lesions (25,26). However, other research shows that
the stiffness of subchondral bone in OA is actually decreased, due
to a reduced mineral content and an increased porosity (27). Further studies have shown the
phenomenon to be more complex and to involve changes in collagen
content and bone density, which potentially lead to weakened
subchondral trabecular bone (28,29). We found that the subchondral bone
degradation of the TXC+OA group significantly decreased compared to
the OA group, indicating the regulation of tide-mark replication by
affecting the arrangement of subchondral bone collagen fibers and
calcium phosphate crystals is one of the mechanisms by which TXC
may be effective in the treatment of OA.
Cell death can be classified according to
enzymological criteria, morphological appearance, immunological
characteristics or functional aspects. Based on morphological
criteria, three types of cell death can be defined; necrosis,
apoptosis and autophagy. Autophagy is a lysosomal degradation
pathway that is essential for differentiation, survival,
homeostasis and development. However, in certain physiological and
pathological conditions, autophagy can also result in a form of
cell death that is termed type II programmed cell death; the
morphological hallmark of autophagy forms the sequestering vesicle,
the autophagosome, which fuses with lysosomes and degrades and
recycles cellular components (30). Atg genes regulate the autophagy
process resulting in the induction and nucleation of autophagic
vesicles, their expansion and fusion with lysosomes, and the
enzymatic degradation and recycling (31,32). Among the Atg genes, Atg1, Atg5,
Atg6 and Atg8 (ULK1, Beclin1 and LC3 in mammals, respectively) are
four major regulators of the autophagy pathway. ULK1 is an
important intermediate in the transduction of pro-autophagic
signals to the formation of autophagosomes (33). Beclin1 forms a complex with Vps34
and type III PI3 kinase, and then allows nucleation of the
autophagic vesicle (34,35). Finally, the formation and
expansion of autophagosome demands two protein conjugation systems
including Atg12 and Atg proteins LC3 (36).
LC3 is the existence of two forms, LC3 II bound to
the autophagosome membrane and LC3 I in the cytoplasm. During the
autophagy process, LC3 I is converted to LC3 II through lipidation
by a ubiquitin-like system leading to the association of LC3 II
with autophagy vesicles. Therefore, the amount of LC3 II is
correlated with the extent of autophagosome formation (12,37). The present results showed that
autophagy was decreased in papain-induced OA, and the feature of
papain-induced cartilage degradation increased cell death
suggesting that loss of autophagy may contribute to cell death. Our
results also showed that the protein of LC3 I/II, ULK1 and Beclin1
in osteoarthritic cartilage was significantly decreased, and TXC
could increase these autophagy markers, demonstrating activation of
autophagy is one of the mechanisms by which TXC inhibits the
papain-induced cartilage degradation.
In summary, the protective role of autophagy in
endochondral ossification was further supported by observations
that its inactivation leads to severe skeletal abnormalities, due
in part to cell death (38). This
study confirmed that autophagy may be a protective or homeostatic
mechanism in normal cartilage. TXC could enhance chondrocyte
autophagy by promoting the expression of LC3 I/II, ULK1 and Beclin1
to inhibit the tidemark replication and cartilage degradation in
the papain-induced OA. These results suggested that compromised
autophagy may represent a novel avenue to delay the development of
OA. Since autophagy can serve to delay the onset of apoptosis,
further experiments are in progress to explore the mechanism of TXC
in the regulation of the relationship between the induction of
autophagy and apoptosis.
Acknowledgements
This study was supported by the
National Natural Science Foundation of China (Grant no. 81102609),
the Key Project of Fujian Provincial Department of Science and
Technology (Grant no. 2012Y0046), the Natural Science Foundation of
Fujian Province (Grant no. 2011J05074) and the Young Talent
Scientific Research Project of Fujian Province Universities (Grant
no. JA12165).
References
|
1
|
Li X, Ye H, Yu F, et al: Millimeter wave
treatment promotes chondrocyte proliferation via G1/S
cell cycle transition. Int J Mol Med. 29:823–831. 2012.PubMed/NCBI
|
|
2
|
Wu MX, Li XH, Lin MN, et al: Clinical
study on the treatment of knee osteoarthritis of shensui
insufficiency syndrome type by electroacupuncture. Chin J Integr
Med. 16:291–297. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Galois L, Etienne S, Grossin L, et al:
Dose-response relationship for exercise on severity of experimental
osteoarthritis in rats: a pilot study. Osteoarthritis Cartilage.
12:779–786. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Panicker S, Borgia J, Fhied C, Mikecz K
and Oegema TR: Oral glucosamine modulates the response of the liver
and lymphocytes of the mesenteric lymph nodes in a papain-induced
model of joint damage and repair. Osteoarthritis Cartilage.
17:1014–1021. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Eswaramoorthy R, Chang CC, Wu SC, Wang GJ,
Chang JK and Ho ML: Sustained release of PTH(1-34) from PLGA
microspheres suppresses osteoarthritis progression in rats. Acta
Biomater. 8:2254–2262. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bonde HV, Talman ML and Kofoed H: The area
of the tidemark in osteoarthritis - a three-dimensional
stereological study in 21 patients. APMIS. 113:349–352. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lyons TJ, Stoddart RW, McClure SF and
McClure J: The tidemark of the chondro-osseous junction of the
normal human knee joint. J Mol Histol. 36:207–215. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Dequeker J, Mokassa L, Aerssens J and
Boonen S: Bone density and local growth factors in generalized
osteoarthritis. Microsc Res Tech. 37:358–371. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Del Carlo M Jr and Loeser RF: Cell death
in osteoarthritis. Curr Rheumatol Rep. 10:37–42. 2008.PubMed/NCBI
|
|
10
|
Almonte-Becerril M, Navarro-Garcia F,
Gonzalez-Robles A, Vega-Lopez MA, Lavalle C and Kouri JB: Cell
death of chondrocytes is a combination between apoptosis and
autophagy during the pathogenesis of Osteoarthritis within an
experimental model. Apoptosis. 15:631–638. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kim HA, Lee YJ, Seong SC, Choe KW and Song
YW: Apoptotic chondrocyte death in human osteoarthritis. J
Rheumatol. 27:455–462. 2000.
|
|
12
|
Caramés B, Taniguchi N, Otsuki S, Blanco
FJ and Lotz M: Autophagy is a protective mechanism in normal
cartilage, and its aging-related loss is linked with cell death and
osteoarthritis. Arthritis Rheum. 62:791–801. 2010.PubMed/NCBI
|
|
13
|
Lotz MK and Caramés B: Autophagy and
cartilage homeostasis mechanisms in joint health, aging and OA. Nat
Rev Rheumatol. 7:579–587. 2011.PubMed/NCBI
|
|
14
|
Li XH, Wu MX, Ye HZ, et al: Experimental
study on the suppression of sodium nitroprussiate-induced
chondrocyte apoptosis by Tougu Xiaotong Capsule-containing serum.
Chin J Integr Med. 17:436–443. 2011. View Article : Google Scholar
|
|
15
|
Kellgren JH and Lawrence JS: Radiological
assessment of rheumatoid arthritis. Ann Rheheum Dis. 16:485–493.
1957. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mankin HJ, Dorfman H, Lippiello L and
Zarins A: Biochemical and metabolic abnormalities in articular
cartilage from osteoarthritic human hips. II. Correlation of
morphology with biochemical and metabolic data. J Bone Joint Surg
Am. 53:523–537. 1971.PubMed/NCBI
|
|
17
|
Klop C, de Vries F, Lalmohamed A, et al:
COX-2-selective NSAIDs and risk of hip or knee replacements: a
population-based case-control study. Calcif Tissue Int. 91:387–394.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zoeger N, Roschger P, Hofstaetter JG, et
al: Lead accumulation in tidemark of articular cartilage.
Osteoarthritis Cartilage. 14:906–913. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gannon FH and Sokoloff L: Histomorphometry
of the aging human patella: histologic criteria and controls.
Osteoarthritis Cartilage. 7:173–181. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Meirer F, Pemmer B, Pepponi G, et al:
Assessment of chemical species of lead accumulated in tidemarks of
human articular cartilage by X-ray absorption near-edge structure
analysis. J Synchrotron Radiat. 18:238–244. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Otterness IG, Chang M, Burkhardt JE,
Sweeney FJ and Milici AJ: Histology and tissue chemistry of
tidemark separation in hamsters. Vet Pathol. 36:138–145. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Pan J, Wang B, Li W, et al: Elevated
cross-talk between subchondral bone and cartilage in osteoarthritic
joints. Bone. 51:212–217. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lacourt M, Gao C, Li A, et al:
Relationship between cartilage and subchondral bone lesions in
repetitive impact trauma-induced equine osteoarthritis.
Osteoarthritis Cartilage. 20:572–583. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bobinac D, Spanjol J, Zoricic S and Maric
I: Changes in articular cartilage and subchondral bone
histomorphometry in osteoarthritic knee joints in humans. Bone.
32:284–290. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Burr DB: Increased biological activity of
subchondral mineralized tissues underlies the progressive
deterioration of articular cartilage in osteoarthritis. J
Rheumatol. 32:1156–1158. 2005.PubMed/NCBI
|
|
26
|
Botter SM, Glasson SS, Hopkins B, et al:
ADAMTS5−/− mice have less subchondral bone changes after induction
of osteo-arthritis through surgical instability: implications for a
link between cartilage and subchondral bone changes. Osteoarthritis
Cartilage. 17:636–645. 2009.
|
|
27
|
Day JS, Ding M, van der Linden JC, Hvid I,
Sumner DR and Weinans H: A decreased subchondral trabecular bone
tissue elastic modulus is associated with pre-arthritic cartilage
damage. J Orthop Res. 19:914–918. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bennell KL, Creaby MW, Wrigley TV and
Hunter DJ: Tibial subchondral trabecular volumetric bone density in
medial knee joint osteoarthritis using peripheral quantitative
computed tomography technology. Arthritis Rheum. 58:2776–2785.
2008.
|
|
29
|
Aula AS, Töyräs J, Tiitu V and Jurvelin
JS: Simultaneous ultrasound measurement of articular cartilage and
subchondral bone. Osteoarthritis Cartilage. 18:1570–1576. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mizushima N, Levine B, Cuervo AM and
Klionsky DJ: Auto-phagy fights disease through cellular
self-digestion. Nature. 451:1069–1075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Srinivas V, Bohensky J, Zahm AM and
Shapiro IM: Autophagy in mineralizing tissues: microenvironmental
perspectives. Cell Cycle. 8:391–393. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Cajee UF, Hull R and Ntwasa M:
Modification by ubiquitin-like proteins: significance in apoptosis
and autophagy pathways. Int J Mol Sci. 13:11804–11831. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chan EY, Kir S and Tooze SA: siRNA
screening of the kinome identifies ULK1 as a multidomain modulator
of autophagy. J Biol Chem. 282:25464–25474. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Furuya N, Yu J, Byfield M, Pattingre S and
Levine B: The evolutionarily conserved domain of Beclin 1 is
required for Vps34 binding, autophagy and tumor suppressor
function. Autophagy. 1:46–52. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Abounit K, Scarabelli TM and McCauley RB:
Autophagy in mammalian cells. World J Biol Chem. 3:1–6. 2012.
|
|
36
|
Ohsumi Y and Mizushima N: Two
ubiquitin-like conjugation systems essential for autophagy. Semin
Cell Dev Biol. 15:231–236. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kabeya Y, Mizushima N, Ueno T, et al: LC3,
a mammalian homologue of yeast Apg8p, is localized in autophagosome
membranes after processing. EMBO J. 19:5720–5728. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Settembre C, Arteaga-Solis E, McKee MD, et
al: Proteoglycan desulfation determines the efficiency of
chondrocyte autophagy and the extent of FGF signaling during
endochondral ossification. Genes Dev. 22:2645–2650. 2008.
View Article : Google Scholar
|