Introduction
Pulmonary hypertension (PH) is a progressive disease
of various origins that results in right heart dysfunction. In all
its variant presentations, this disease is estimated to affect up
to 100 million people worldwide, and is associated with a poor
prognosis (1). PH can be
idiopathic or associated with other diseases, such as connective
tissue diseases, congenital heart defects, portal hypertension,
left heart disease and chronic obstructive pulmonary disease
(COPD), or it can occur after long-term living in plateau
environment (1). Although the
exact pathogenesis of PH remains unclear, there is increasing
support that pulmonary artery endothelial cell (PAEC) apoptosis
induced by a variety of factors, including hypoxia, is the initial
step and triggering event for PH (2–4).
PAEC loss results in an abnormal overgrowth of pulmonary artery
smooth muscle cells, provides conditions favoring the emergence of
apoptosis-resistant hyperproliferative endothelial cells (ECs), and
finally leads to advanced PH (3–5).
Current therapies for chronic PH include prostanoids, endothelin
receptor antagonists and phosphodiesterase inhibitors; these
therapeutic approaches mainly provide symptomatic relief and slow
down disease progression (6).
There is, therefore, a genuine need for novel treatments that
prevent progression of PH by interfering with the pathogenesis of
the disease at the initial stage. To this end, antagonism of human
PAEC (HPAEC) apoptosis may be a useful approach on which to base
novel treatments for PH.
Nicorandil, which is generally used for the
management of angina pectoris, has a unique pharmacological profile
that includes a nitrate-like effect to dilate peripheral coronary
arteries, and an action as an agonist at mitochondrial adenosine
triphosphate-sensitive potassium (mitoKATP) channels. In
some cell types, nicorandil has been shown to inhibit apoptosis
induced by a number of factors, through activation of
mitoKATP channels (7–12).
However, whether nicorandil has an anti-apoptotic effect in HPAECs
remains unknown.
Emerging evidence has indicated an important role
for endothelial nitric oxide synthase (eNOS) in the regulation of
EC apoptosis. Reduced expression of eNOS leads to a decreased NO
synthesis which is a primary factor in the pathophysiology of PH
(13). In monocrotaline
(MCT)-induced PH rats, nicorandil has been reported to exert a
beneficial effect through an increase in eNOS expression in heart
and lung tissue (14,15). Furthermore, some studies have
found that the opening of mitoKATP channels could induce
eNOS expression (16,17). However, it is currently unclear
whether nicorandil has the ability to enhance eNOS expression via
activation of mitoKATP channels in HPAECs.
Nuclear factor-κB (NF-κB) is a transcription factor
that plays an important role in the regulation of the inflammatory
response and apoptosis in several cells types (18). Recent studies have observed an
increased expression of NF-κB in PH, and have shown that blockade
of NF-κB activity could reduce the development of PH (19–21). Of note, nicorandil has been
reported to reduce myocardial reperfusion injury by attenuating
NF-κB activation (22), and
levosimendan, a KATP channel opener, has been found to
inhibit NF-κB expression in human umbilical vein endothelial cells
(HUVECs) (23). However, little
data exists concerning the effect of nicorandil on the NF-κB
pathway in HPAECs. Furthermore, although there is accumulating
evidence that eNOS or NO can mediate inhibition of NF-κB signaling
and suppress NF-κB-dependent apoptosis and inflammation (24–26), little is known about the
relationship between eNOS and NF-κB in HPAECs.
The aim of the present study was to determine
whether treatment with nicorandil attenuates apoptosis of HPAECs
exposed to hypoxia, and, if so, to investigate the mechanisms
involved.
Materials and methods
Reagents
Nicorandil was purchased from Tokyo Chemical
Industry Co., Ltd. (Tokyo, Japan). Endothelial Cell Medium (ECM),
vascular endothelial growth factor (VEGF), fetal bovine serum
(FBS), penicillin and streptomycin were obtained from HyClone
Laboratories, Inc. (Logan, UT, USA). Antibodies against Bax, Bcl-2,
caspase-3 and -9, eNOS, NF-κB and IκBα were purchased from Cell
Signaling Technology (Beverly, MA, USA), and antibody against
β-actin was purchased from Santa Cruz Biotechnology, Inc. (Santa
Cruz, CA, USA). 5-Hydroxydecanoate (5-HD), NG-nitro-L-arginine
methyl ester (L-NAME) and pyrrolidine dithiocarbamate (PDTC) were
purchased from Sigma (St. Louis, MO, USA).
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT),
Hoechst 33342, Annexin V-fluorescein isothiocyanate (FITC) and
propidium iodide (PI) were obtained from Sigma. AnaeroPack-Anaero
was purchased from Mitsubishi Gas Chemical Co. (Tokyo, Japan). All
other chemicals were of the highest purity available
commercially.
Cell culture and treatment
HPAECs were obtained from ScienCell Research
Laboratories (San Diego, CA, USA). HPAECs (1×105/75
cm2 flask) were seeded in ECM containing 10% (v/v) FBS,
1% (v/v) VEGF, 100 U/ml penicillin and 100 U/ml streptomycin, at
37°C in a 5% CO2-humidified atmosphere. The culture
medium was replaced every 2–3 days until 80–90% confluence was
achieved. HPAECs of the 3rd to 5th passages, in an actively growing
condition, were used for the experiments. HPAECs were divided into
6 treatment groups: i) control group: cells were cultured under
normoxic conditions for 24 h (20% O2, 5%
CO2); ii) hypoxia group: cells were cultured in the
hypoxic chamber using AnaeroPack-Anaero, a disposable
oxygen-absorbing and CO2-generating agent, for 24 h;
iii) hypoxia + nicorandil group: cells were pretreated with
nicorandil (100 μM), and then cultured in the hypoxic
chamber using AnaeroPack-Anaero for 24 h; iv) hypoxia + nicorandil
+ 5-HD group: cells were pretreated with nicorandil (100 μM)
and 5-HD (500 μM), and then cultured in the hypoxic chamber
using AnaeroPack-Anaero for 24 h; v) hypoxia + nicorandil + L-NAME
group: cells were pretreated with nicorandil (100 μM) and
L-NAME (300 μM), and then cultured in the hypoxic chamber
using AnaeroPack-Anaero for 24 h; vi) hypoxia + PDTC group: cells
were pretreated with PDTC (10 μM), and then cultured in the
hypoxic chamber using AnaeroPack-Anaero for 24 h. The
concentrations of nicorandil, 5-HD, PDTC and L-NAME used in this
study were selected on the basis of previously published studies
(8,27,28). 5-HD, L-NAME and PDTC were added to
the medium 30 min prior to the addition of nicorandil. The
AnaeroPack started to absorb oxygen within 1 min, oxygen tension
inside the box dropped to 1 mmHg within 1 h (O2 <1%,
CO2 ~5%).
MTT cell viability assay
Cell viability was determined by MTT incorporation.
HPAECs were seeded in 96-well culture plates, cultivated, and
divided into different treatment groups as described above.
Following addition of the appropriate drugs, 20 μl assay
medium containing 5 mg/ml MTT was added to each well before the
culture was terminated. After 4 h of incubation at 37°C, the medium
was aspirated and the cells were lysed by the addition of 100
μl DMSO. Following a 10-min incubation at 37°C, the optical
density of each sample was measured in an ELISA microplate reader
using test and reference wavelengths of 570 nm.
Hoechst 33342 staining assay
HPAECs were seeded in 24-well culture plates,
cultivated, and divided into different treatment groups as
described above. After the addition of appropriate drugs, cells
were washed twice with PBS and treated with 100 μl Hoechst
33342 (10 μg/ml) for 15 min at 37°C, in the dark. The medium
was aspirated and samples were then analyzed by an Olympus
fluorescence microscopy using an excitation wavelength of 346 nm
and an emission wavelength of 460 nm.
Annexin V-PI analysis by flow
cytometry
Following exposure to the appropriate drugs, cells
in the various treatment groups were harvested and resuspended in
PBS at a density of 1×106 cells/ml. Following
centrifugation at 1,000 × g for 5 min, 195 μl
FITC-conjugated Annexin V binding buffer and 5 μl Annexin
V-FITC were added. Following gentle vortexing, the mixture was
incubated for 15 min at room temperature, in the dark. Following
centrifugation at 1,000 × g for 5 min, the medium was aspirated and
190 μl FITC-conjugated Annexin V binding buffer and 10
μl PI were added. Immediately after gentle vortexing, the
sample was analyzed by flow cytometry. Early apoptotic cells were
characterized by high Annexin V binding and low PI staining,
whereas late apoptotic and necrotic cells stained strongly with
both Annexin V and PI.
Western blot analysis
Cells were lysed in iced lysis buffer (97.9% RIPA,
1% PMSF, 1% phosphatase inhibitors and 0.1% protease inhibitors).
Total protein (50 μg/lane) was separated by SDS-PAGE and
transferred to a PVDF membrane (Millipore, Billerica, MA, USA).
Following incubation in blocking solution (5% nonfat milk),
membranes were incubated overnight at 4°C with primary antibodies
against Bax, Bcl-2, caspase-3 and -9, eNOS, NF-κB, IκBα or β-actin.
Membranes were then washed. β-actin was detected directly with
enhanced chemiluminescence reagents. The other proteins of interest
were detected with enhanced chemiluminescence reagents following
incubation with horseradish peroxidase-conjugated secondary
antibody (1:8,000 dilution) for 1 h. The relative density of each
protein band was normalized to that of β-actin. All results were
representative of at least three independent experiments.
Statistical analysis
Data are presented as the means ± SD, derived from
at least three independent experiments. Statistical significance
between groups was analyzed by one-way ANOVA followed by Tukey or
Dunnett’s T3 test, as appropriate, using SPSS 15.0 software. A
value of P<0.05 was considered to indicate statistically
significant differences.
Results
Effect of nicorandil on hypoxia-induced
loss of HPAEC viability
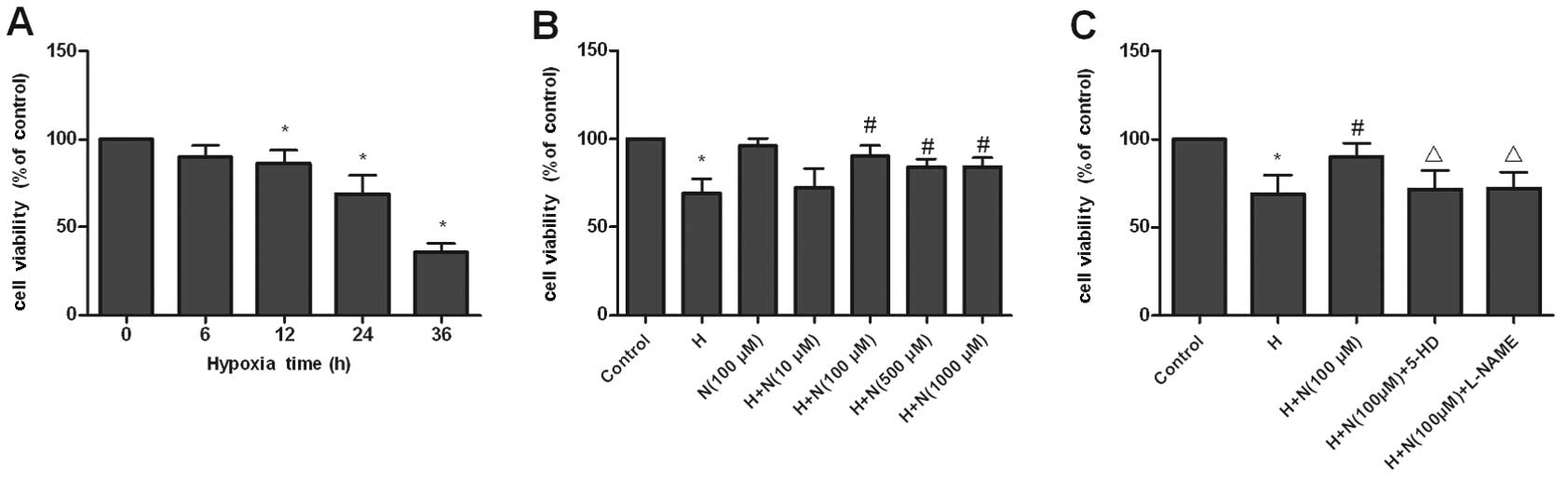
To determine whether hypoxia can reduce HPAEC
viability, HPAECs were cultured under hypoxic conditions for 0, 6,
12, 24 or 36 h, and cell viability was analyzed using MTT. HPAEC
viability following hypoxia for 6 or 12 h was not significantly
different (P>0.05) to that of control cells not exposed to
hypoxia (Fig. 1A). However,
hypoxia for 24 or 36 h decreased HPAEC viability to 67.5±3.5 and
36.7±2.1%, respectively, of the control group (P<0.05) (Fig. 1A). Based on these data, subsequent
experiments were carried out using hypoxia for 24 h, unless
otherwise stated. Three different concentrations (100, 500 and
1,000 μM) of nicorandil improved cell viability. Nicorandil
(100 μM) itself did not have a significant effect on cell
viability (Fig. 1B). However, the
protective effects of nicorandil against hypoxia-induced decreases
in cell viability were blunted by 5-HD or L-NAME (Fig. 1C); under hypoxic conditions, the
reduction in cell viability in the presence of 5-HD or L-NAME
(combined with nicorandil) was significantly greater (P<0.05)
than that in their absence (i.e., nicorandil alone). In addition,
nicorandil, 5-HD or L-NAME alone was without effect on HPAEC
viability under normoxic conditions (data not shown). These results
demonstrated that nicorandil was able to protect HPAECs against
decreased cell viability induced by hypoxia, whereas 5-HD or L-NAME
was found to inhibit these beneficial effects of nicorandil.
Effect of nicorandil on hypoxia-induced
apoptosis of HPAECs
To investigate whether the protective effect of
nicorandil on hypoxia-induced cytotoxicity may be due to the
attenuation of hypoxia-induced apoptosis, HPAEC apoptosis was
determined using a Hoechst 33342 staining assay and flow
cytometry.
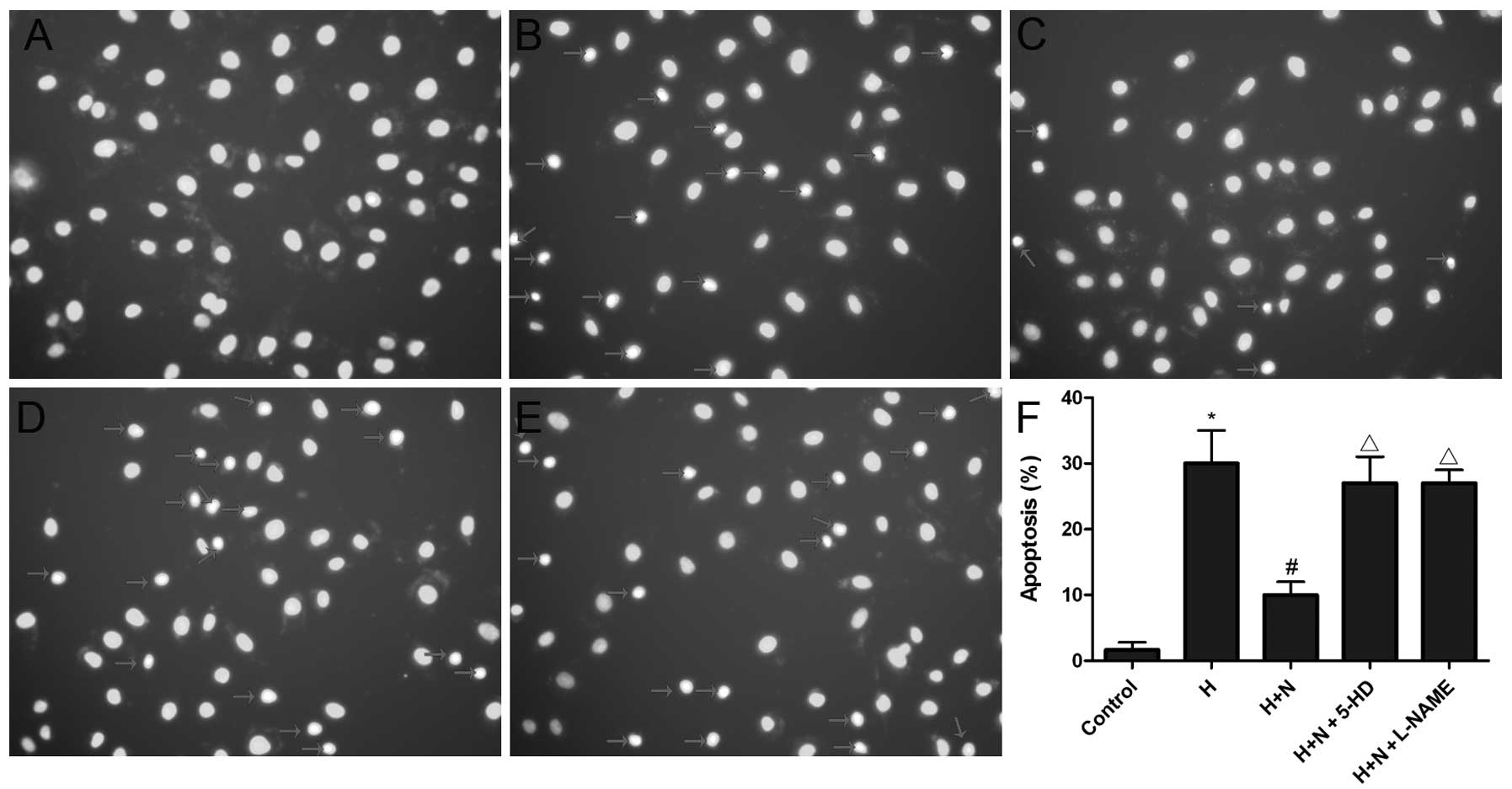
The Hoechst 33342 staining assay demonstrated that
exposure to hypoxia for 24 h increased the apoptotic ratio of
cells, which showed a profile of cell shrinkage, chromatin
condensation and fragmented fluorescent nuclei (Fig. 2). The number of Hoechst
33342-positive cells and the apoptotic ratio were significantly
reduced by nicorandil (Fig. 2).
However, the protective effects of nicorandil against
hypoxia-induced cell apoptosis were blunted by 5-HD or L-NAME, as
shown by the increase in the number of Hoechst 33342-positive cells
and in the apoptotic ratio in the presence of 5-HD or L-NAME,
compared with the hypoxia + nicorandil group (Fig. 2).
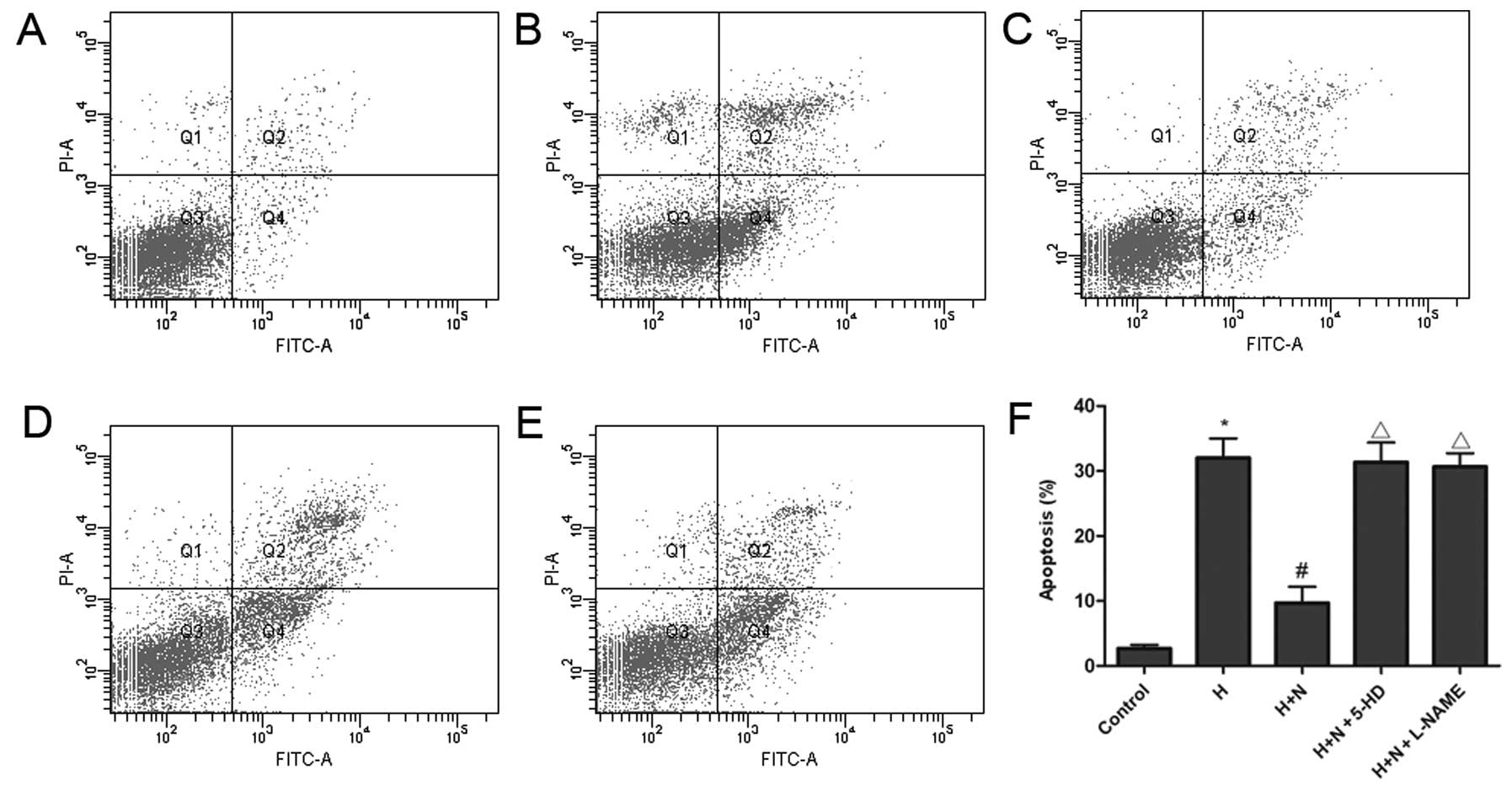
These findings were supported by the results of
experiments using Annexin V and PI, analyzed by flow cytometry.
Exposure to hypoxia for 24 h induced a significant increase in the
number of apoptotic cells, as identified by flow cytometry,
compared with normoxic culture (Fig.
3). Nicorandil markedly promoted cell survival. However,
pretreatment with 5-HD or L-NAME abolished the anti-apoptotic
effect of nicorandil in cultured HPAECs exposed to hypoxia.
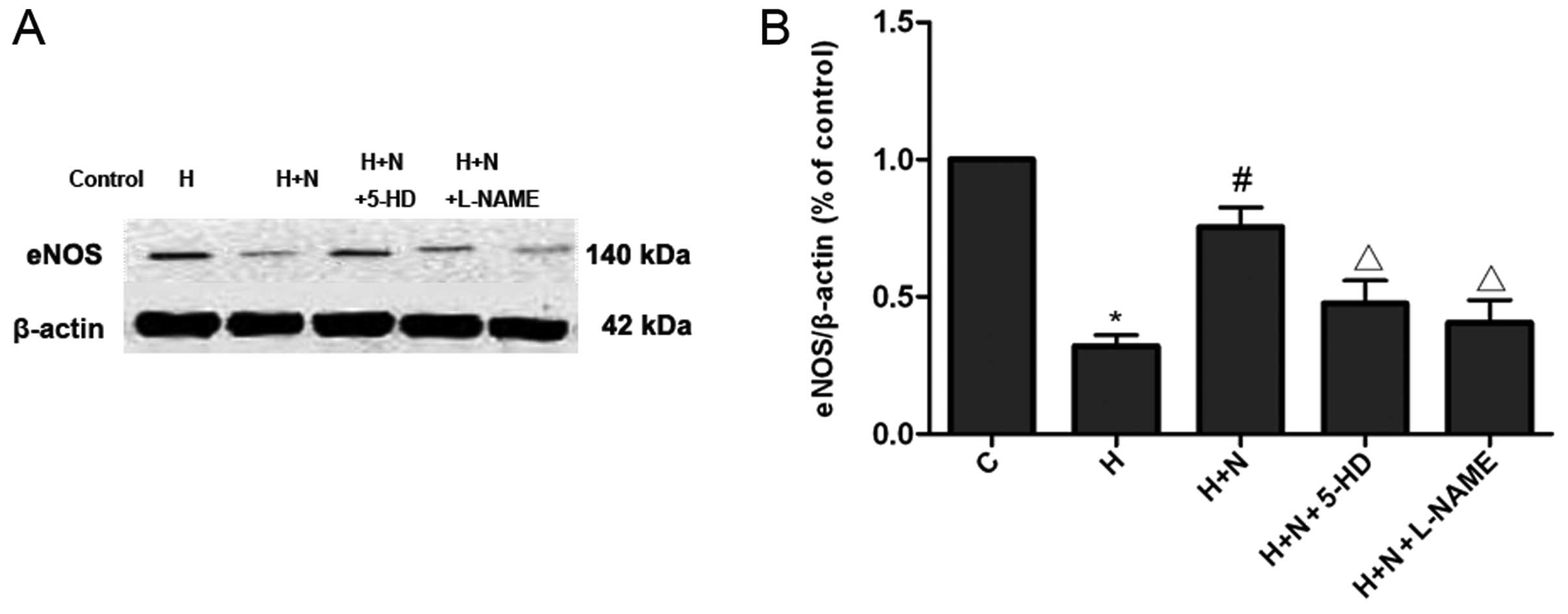
Effect of nicorandil on activation of
eNOS
To further investigate the mechanism underlying the
anti-apoptotic effect of nicorandil, we examined eNOS protein
expression in the various treatment groups by western blot
analysis. Hypoxia decreased the expression of eNOS protein,
compared with the control group, whereas nicorandil markedly
increased the expression of eNOS compared with the hypoxia group
(Fig. 4). However, the effects of
nicorandil to enhance eNOS expression were suppressed by 5-HD or
L-NAME (Fig. 4). By contrast,
nicorandil, 5-HD and L-NAME alone had no influence on eNOS
expression in HPAECs under normoxic conditions (data not
shown).
Effect of nicorandil on inhibition of the
NF-κB pathway
As NF-κB plays an important role in the regulation
of cell death in some cell types, we examined its effects on
hypoxia-induced HPAEC apoptosis. Hypoxia was found to increase
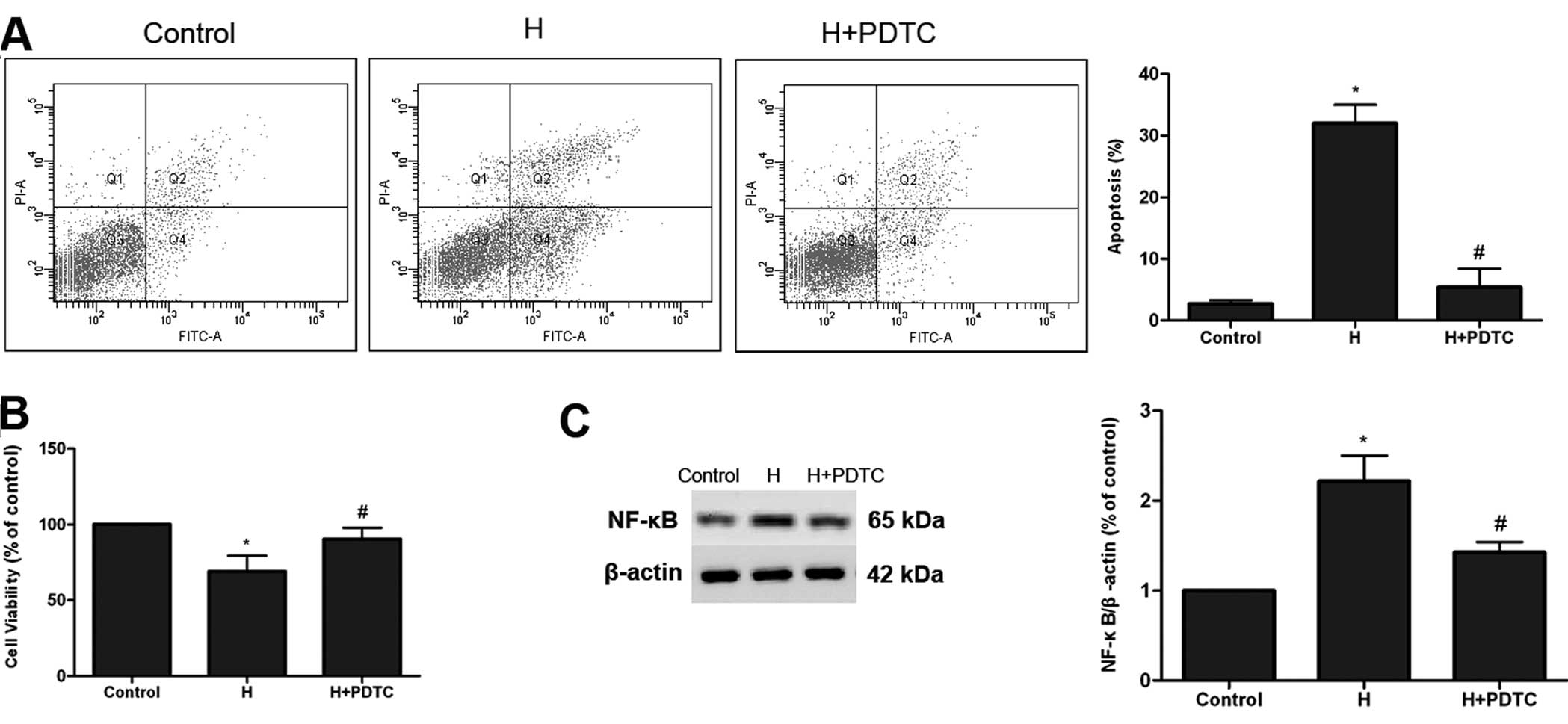
expression of NF-κB protein compared to the control group (Fig. 5C), whereas pretreatment with PDTC
markedly downregulated the expression of NF-κB (Fig. 5C), inhibited hypoxia-induced cell
apoptosis (Fig. 5A) and restored
decreased HPAEC viability (Fig.
5B). By contrast, PDTC alone had no influence on HPAEC
apoptosis and viability under normoxic conditions (data not shown).
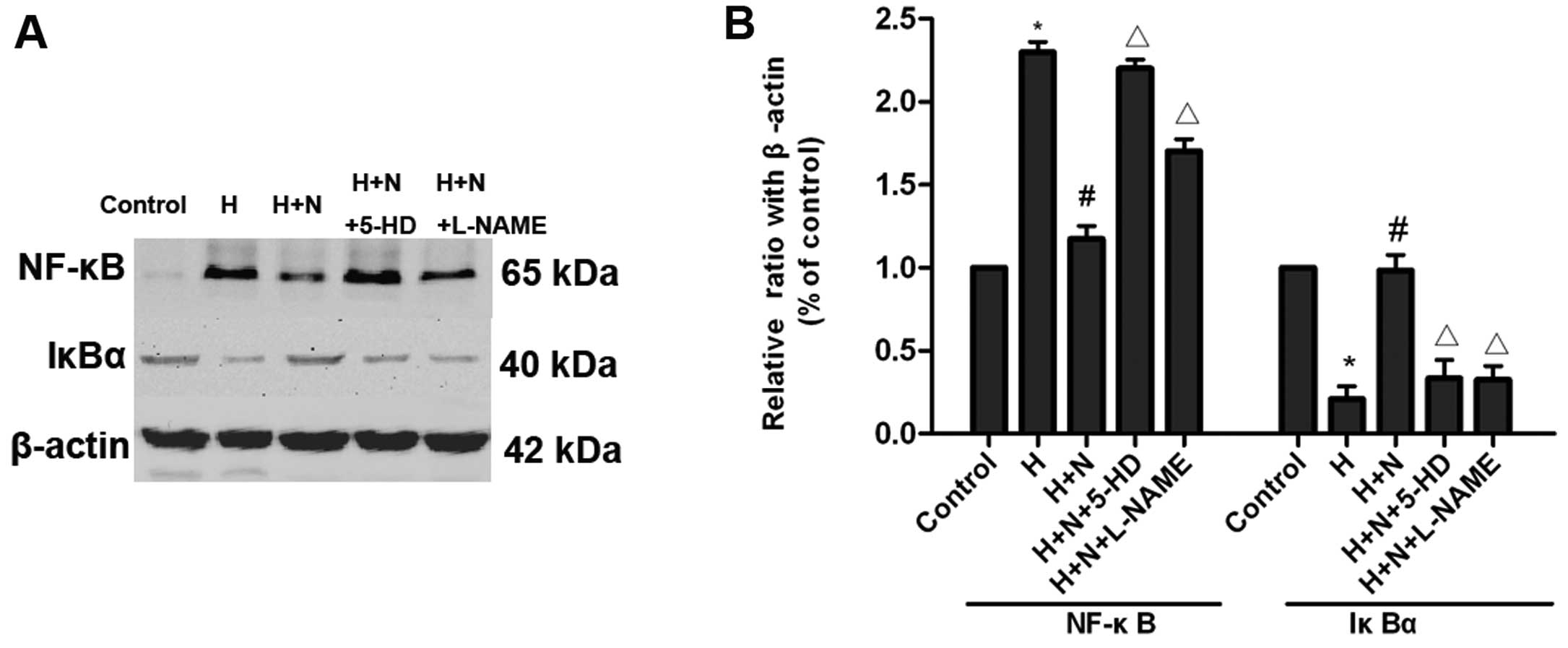
We next examined the effect of nicorandil on the regulation of the
expression of NF-κB and IκBα (a protein that inhibits NF-κB), and
found that nicorandil markedly decreased the expression of NF-κB
and increased the expression of IκBα, compared with the hypoxia
group. However, these effects of nicorandil were reduced by 5-HD or
L-NAME (Fig. 6).
Effect of nicorandil on caspase-3 and -9
protein expression, and the Bax/Bcl-2 protein expression ratio
To explore the potential apoptotic signals involved
in the protective effect of nicorandil on hypoxia-induced cell
death, we examined protein expression levels of Bax, Bcl-2,
caspase-3 and -9 in the various treatment groups, using western
blot analysis. Hypoxia increased the Bax/Bcl-2 protein expression
ratio, as well as caspase-3 and -9 protein levels, compared with
the control group (Fig. 7).
Nicorandil markedly decreased the Bax/Bcl-2 protein expression
ratio as well as caspase-3 and -9 protein expression, compared with
the hypoxia group (Fig. 7).
However, these effects of nicorandil were reduced by 5-HD or L-NAME
(Fig. 7).
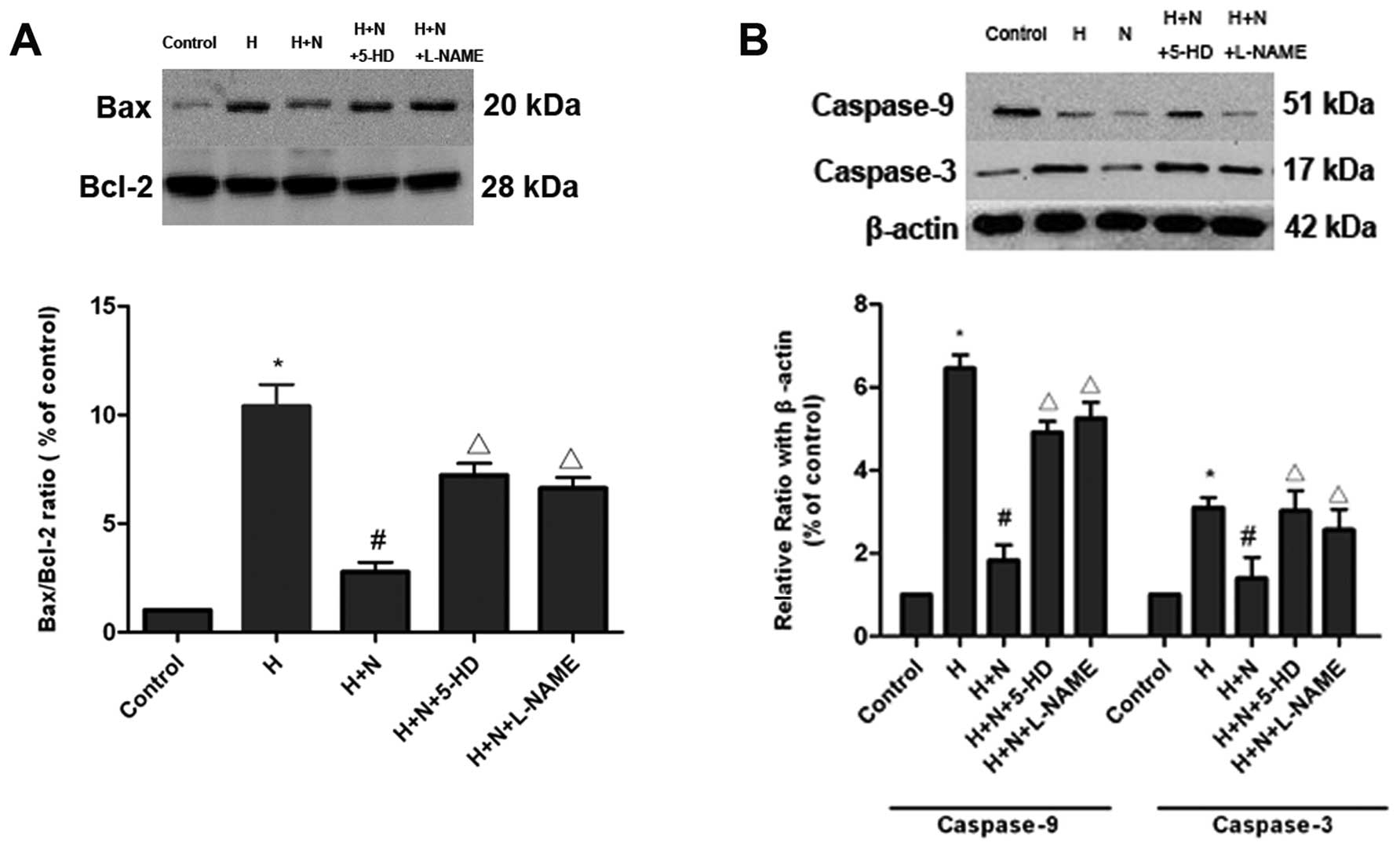 | Figure 7.Effect of nicorandil on the increased
Bax/Bcl-2 expression ratio, and enhanced caspase-3 and -9
expression, induced by hypoxia. (A) Representative protein
expression of Bax and Bcl-2 in each treatment group, assayed by
western blotting, with densitometric analysis of the corresponding
bands. (B) Representative protein expression of caspase-9 and -3 in
each group, assayed by western blotting, with densitometric
analysis of the corresponding bands. Data are mean ± SD values from
three independent experiments. *P<0.05 vs. control
group; #P<0.05 vs. hypoxia group;
▵P<0.05 vs. hypoxia+nicorandil group. H, hypoxia (24
h); N, nicorandil (100 μM); 5-HD, 5-hydroxydecanoate (500
μM); L-NAME, NG-nitro-L-arginine methyl ester (300
μM). |
Discussion
There are two major findings in the present study
that expand our understanding of the mechanisms of action of
nicorandil in HPAECs. First, we found that nicorandil protected
HPAECs from hypoxia-induced apoptosis. Second, we demonstrated that
the anti-apoptotic effect of nicorandil was through activation of
mitoKATP channels and increased eNOS expression, with
subsequent inhibition of the NF-κB pathway and the mitochondrial
apoptotic pathway.
HPAEC apoptosis has been proposed to be the initial
step and the triggering event for PH, including hypoxic PH (HPH)
(2–4), and chronic hypoxia is known to be an
important cause of EC apoptosis. Date et al (8) showed that nicorandil could inhibit
serum starvation-induced apoptosis in vascular endothelial cells
through activation of mitoKATP channels, and a similar
result was found in oxidative stress-induced apoptosis in neurons
(9). In the present study, we
demonstrated that nicorandil markedly inhibited the decreased HPAEC
viability and increased HPAEC apoptosis induced by hypoxia.
However, this beneficial effect of nicorandil was almost completely
suppressed by pretreatment with 5-HD, a mitoKATP channel
inhibitor, indicating that the effect of nicorandil on HPAECs is
mediated via activation of mitoKATP channels.
eNOS, an important enzyme in the production of NO,
is present in vascular endothelial cells, and a decreased
expression level or abnormality of eNOS protein has been shown to
cause EC apoptosis in the early stage of PH (including HPH),
leading to the development of PH (13,29–31). For example, mutant mice lacking
the eNOS gene, or newborn lambs treated with an eNOS inhibitor,
were found to develop progressive elevation of pulmonary arterial
pressure and resistance (30,31), and individuals with PH have been
shown to have reduced levels of pulmonary arterial eNOS expression
(13). Hongo et al
(14) reported that, in an
MCT-induced PH model, nicorandil was able to protect endothelial
function and enhance eNOS expression, and a similar result was
found in cardiac tissue (15).
Furthermore, Grossini et al (16) showed that opening
mitoKATP channels could induce eNOS expression through
Akt, ERK and p38 activation, and Roth et al (17) observed that ischemic
preconditioning could open mitoKATP channels and
subsequently activate eNOS. In our study, we found that
pretreatment with nicorandil could restore decreased expression of
eNOS induced by hypoxia in HPAECs, while this effect could be
inhibited by 5-HD and L-NAME, an eNOS inhibitor, indicating that
the effect of nicorandil to enhance eNOS expression was via
activation of mitoKATP.
NF-κB can be activated in a variety of cells in
response to various factors, and can thereby regulate cell
apoptosis (27,32,33). IκBα is the inhibitory protein of
NF-κB, and degradation of IκBα results in NF-κB activation.
Matsushita et al (32)
showed that hypoxia-induced endothelial apoptosis was through
NF-κB-mediated Bcl-2 suppression. Similarly, Chen et al
(33) reported that HUVECs
treated with intermittent high glucose showed activation of the
NF-κB pathway, which inhibited cell proliferation and induced cell
apoptosis. Furthermore, Huang et al (34) found that NF-κB played a pivotal
role in disrupting PAEC membrane integrity in an MCT-induced PH
model. Our present study is consistent with previous studies in
showing that hypoxia upregulated the expression of NF-κB protein
and increased apoptosis of HPAECs, while PDTC, an inhibitor of
NF-κB, significantly reduced the enhanced apoptosis of HPAECs and
the increased expression of NF-κB. There have also been some
reports of an interaction between NF-κB and mitoKATP
channels. For example, Kawamura et al (22) reported that nicorandil could
attenuate NF-κB activation via opening of mitoKATP
channels, and thereby reduce myocar-dial reperfusion injury. In
addition, Revermann et al (23) demonstrated that activation of
KATP channels could inhibit NF-κB expression in HUVECs.
However, whether nicorandil could inhibit the NF-κB pathway via
activation of mitoKATP was previously unknown. Our data
indicated that nicorandil markedly decreased the protein expression
of NF-κB, while increasing the expression of IκBα in HPAECs, and
that this effect was diminished by 5-HD. This, therefore, provides
the first evidence that nicorandil can inhibit the NF-κB pathway in
HPAECs via activation of mitoKATP.
We therefore further explored the link between eNOS
and NF-κB. A study on HPAECs found that NO could inhibit
hyperoxia-induced NF-κB activation (25). Moreover, Greco et al
(35) reported that inhibiting
the expression of eNOS was associated with enhanced NF-κB
activation and a significant increase in infarct volume in a
cerebral ischemia/ reperfusion rat model. A recent study reported
that elevated cardiac NO levels following eNOS gene transfer in
mice led to a decrease in nuclear translocation of NF-κB in a model
of myocardial ischemia (24). Our
study is the first to show that the effects of nicorandil to
inhibit the NF-κB pathway could be diminished by L-NAME in HPAECs,
indicating a relationship between eNOS and NF-κB in these cells,
such that upregulation of eNOS expression could inhibit the NF-κB
pathway.
The apoptotic signals involved in the protective
effect of nicorandil on hypoxia-induced HPAEC death were also
investigated in our study. We had already shown that nicorandil
could act as a mitoKATP channel opener of HPAECs induced
by hypoxia; this result indicated that nicorandil may exert an
influence on the mitochondrial apoptotic pathway. There are at
least two broad pathways that lead to apoptosis, an ‘extrinsic’ and
an ‘intrinsic’ pathway, with apoptosis in mitochondria being the
best known intrinsic apoptotic pathway. Several different causes,
including hypoxia, reduce the expression ratio of Bcl-2/Bax (which
is an important sign of mitochondrial cell apoptosis), leading to
the loss of mitochondrial inner membrane potential (Δψm) and the
triggering of a release of mitochondrial cytochrome c into
the cytosol. There is also activation and upregulation of caspase-9
and -3, and eventually induction of cell apoptosis. Opening
mitoKATP channels can increase Δψm (36), maintain the expression ratio of
Bcl-2/Bax (11) and inhibit cell
apoptosis. In the present study, we found that nicorandil could
inhibit the increased Bax/Bcl-2 expression ratio, and the enhanced
caspase-3 and -9 expression, induced by hypoxia in HPAECs. This
effect of nicorandil could be diminished both by 5-HD and L-NAME,
indicating that the anti-apoptotic effect of nicorandil was through
inhibition of the mitochondrial apoptotic pathway, and that this
action involved an increase in the expression of eNOS via
activation of the mitoKATP channel.
There are some limitations to the present study.
First, whether it is nicorandil’s action as an NO donor that is
involved in the protection of HPAECs against apoptosis remains
unknown. In the current study, the protective effect of nicorandil
could be almost completely inhibited by 5-HD. Furthermore, Harada
et al (37) showed that
nicorandil, acting as an NO donor, stimulated PKC-δ and then opened
mitoKATP channels. This leads us to the hypothesis that
the nitrate-like effect of nicorandil is indeed involved in the
protection against HPAEC apoptosis, although opening of the
mitoKATP channel also contributes to it. Second, our
study has shown that eNOS could mediate enhanced expression of
NF-κB protein; however, whether there is feedback between eNOS and
NF-κB is not known, and further studies are required to clearly
define the relationship between eNOS and NF-κB.
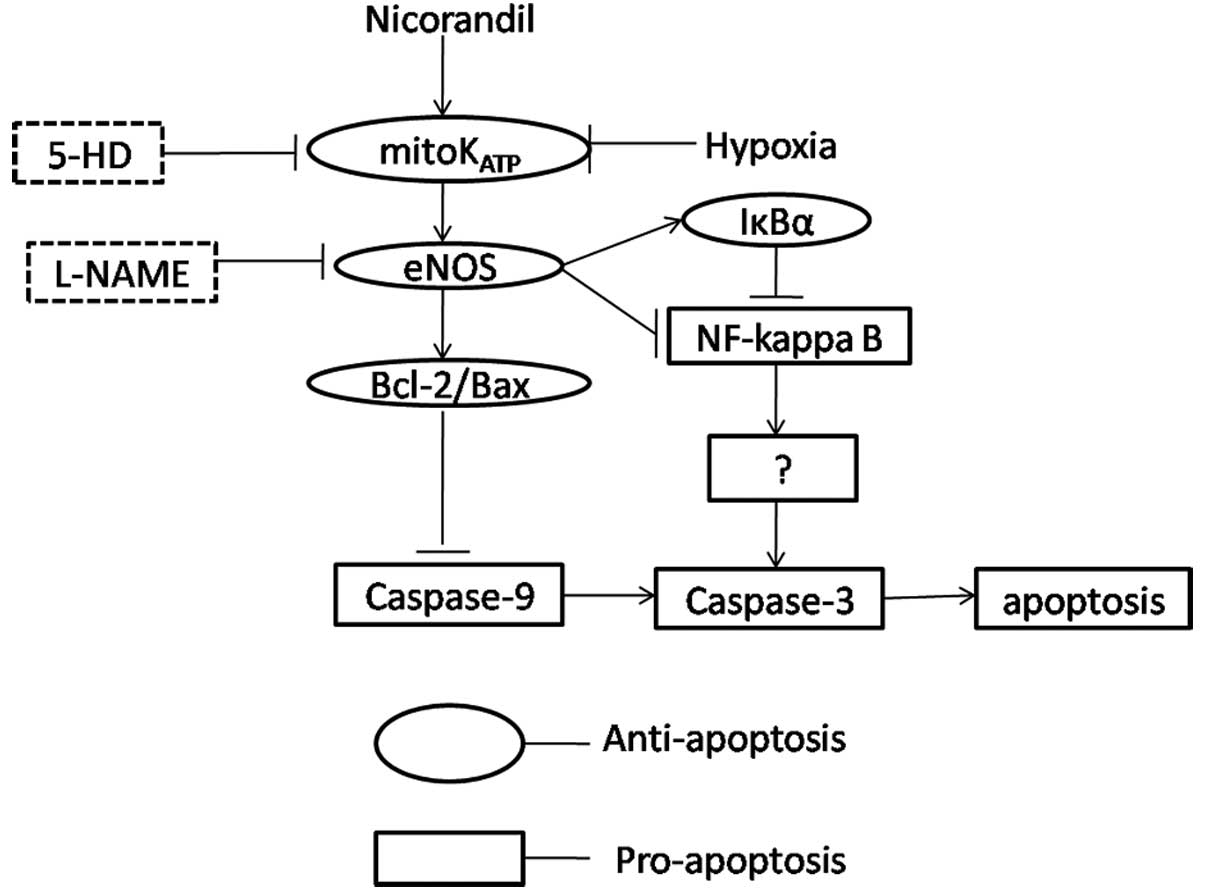
In summary, the present study shows a working
hypothesis that nicorandil suppresses hypoxia-induced apoptosis of
cultured HPAECs through activation of mitoKATP and
increased eNOS expression, which subsequently acts to inhibit the
NF-κB pathway and the mitochondrial apoptotic pathway (Fig. 8). This provides strong evidence
that nicorandil may be useful as an alternative drug for the
treatment of HPH.
Acknowledgements
This study was supported by grants
from the National Natural Science Foundation of China (nos.
81273571 and 81200159), the National Major Scientific and
Technological Special Project for ‘Significant New Drugs
Development’ (2011ZX09302-003-02), the Jiangsu Province Major
Scientific and Technological Special Project (BM2011017), and the
Open Project Program of the key discipline of the Public Health
Department of Jiangsu Province (no. XK13_200902).
References
|
1.
|
Simonneau G, Robbins IM, Beghetti M, et
al: Updated clinical classification of pulmonary hypertension. J Am
Coll Cardiol. 54(Suppl 1): S43–S54. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Jurasz P, Courtman D, Babaie S and Stewart
DJ: Role of apoptosis in pulmonary hypertension: from experimental
models to clinical trials. Pharmacol Ther. 126:1–8. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Sakao S, Taraseviciene-Stewart L, Wood K,
Cool CD and Voelkel NF: Apoptosis of pulmonary microvascular
endothelial cells stimulates vascular smooth muscle cell growth. Am
J Physiol Lung Cell Mol Physiol. 291:L362–L368. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Taraseviciene-Stewart L, Kasahara Y, Alger
L, et al: Inhibition of the VEGF receptor 2 combined with chronic
hypoxia causes cell death-dependent pulmonary endothelial cell
proliferation and severe pulmonary hypertension. FASEB J.
15:427–438. 2001. View Article : Google Scholar
|
|
5.
|
Teichert-Kuliszewska K, Kutryk MJ,
Kuliszewski MA, et al: Bone morphogenetic protein receptor-2
signaling promotes pulmonary arterial endothelial cell survival:
implications for loss-of-function mutations in the pathogenesis of
pulmonary hypertension. Circ Res. 98:209–217. 2006. View Article : Google Scholar
|
|
6.
|
McLaughlin VV and McGoon MD: Pulmonary
arterial hypertension. Circulation. 114:1417–1431. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Nagata K, Obata K, Odashima M, et al:
Nicorandil inhibits oxidative stress-induced apoptosis in cardiac
myocytes through activation of mitochondrial ATP-sensitive
potassium channels and a nitrate-like effect. J Mol Cell Cardiol.
35:1505–1512. 2003. View Article : Google Scholar
|
|
8.
|
Date T, Taniguchi I, Inada K, et al:
Nicorandil inhibits serum starvation-induced apoptosis in vascular
endothelial cells. J Cardiovasc Pharmacol. 46:721–726. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Teshima Y, Akao M, Baumgartner WA and
Marban E: Nicorandil prevents oxidative stress-induced apoptosis in
neurons by activating mitochondrial ATP-sensitive potassium
channels. Brain Res. 990:45–50. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Akao M, Teshima Y and Marban E:
Antiapoptotic effect of nicorandil mediated by mitochondrial
atp-sensitive potassium channels in cultured cardiac myocytes. J Am
Coll Cardiol. 40:803–810. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Nishikawa S, Tatsumi T, Shiraishi J, et
al: Nicorandil regulates Bcl-2 family proteins and protects cardiac
myocytes against hypoxia-induced apoptosis. J Mol Cell Cardiol.
40:510–519. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Zuo XR, Wang Q, Cao Q, et al: Nicorandil
prevents right ventricular remodeling by inhibiting apoptosis and
lowering pressure overload in rats with pulmonary arterial
hypertension. PloS One. 7:e444852012. View Article : Google Scholar
|
|
13.
|
Giaid A and Saleh D: Reduced expression of
endothelial nitric oxide synthase in the lungs of patients with
pulmonary hypertension. N Engl J Med. 333:214–221. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Hongo M, Mawatari E, Sakai A, et al:
Effects of nicorandil on monocrotaline-induced pulmonary arterial
hypertension in rats. J Cardiovasc Pharmacol. 46:452–458. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Horinaka S, Kobayashi N, Higashi T, Hara
K, Hara S and Matsuoka H: Nicorandil enhances cardiac endothelial
nitric oxide synthase expression via activation of adenosine
triphosphate-sensitive K channel in rat. J Cardiovasc Pharmacol.
38:200–210. 2001. View Article : Google Scholar
|
|
16.
|
Grossini E, Molinari C, Caimmi PP, Uberti
F and Vacca G: Levosimendan induces NO production through p38 MAPK,
ERK and Akt in porcine coronary endothelial cells: role for
mitochondrial K(ATP) channel. Br J Pharmacol. 156:250–261. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Roth S, Dreixler JC, Shaikh AR, Lee KH and
Bindokas V: Mitochondrial potassium ATP channels and retinal
ischemic preconditioning. Invest Ophthalmol Vis Sci. 47:2114–2124.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Ghosh S and Karin M: Missing pieces in the
NF-kappaB puzzle. Cell. 109(Suppl): S81–S96. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Kimura S, Egashira K, Chen L, et al:
Nanoparticle-mediated delivery of nuclear factor kappaB decoy into
lungs ameliorates monocrotaline-induced pulmonary arterial
hypertension. Hypertension. 53:877–883. 2009. View Article : Google Scholar
|
|
20.
|
Ortiz LA, Champion HC, Lasky JA, et al:
Enalapril protects mice from pulmonary hypertension by inhibiting
TNF-mediated activation of NF-kappaB and AP-1. Am J Physiol Lung
Cell Mol Physiol. 282:L1209–1221. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Sawada H, Mitani Y, Maruyama J, et al: A
nuclear factor-kappaB inhibitor pyrrolidine dithiocarbamate
ameliorates pulmonary hypertension in rats. Chest. 132:1265–1274.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Kawamura T, Kadosaki M, Nara N, Wei J,
Endo S and Inada K: Nicorandil attenuates NF-kappaB activation,
adhesion molecule expression, and cytokine production in patients
with coronary artery bypass surgery. Shock. 24:103–108. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Revermann M, Schloss M, Mieth A, et al:
Levosimendan attenuates pulmonary vascular remodeling. Intensive
Care Med. 37:1368–1377. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Smith RS Jr, Agata J, Xia CF, Chao L and
Chao J: Human endothelial nitric oxide synthase gene delivery
protects against cardiac remodeling and reduces oxidative stress
after myocardial infarction. Life Sci. 76:2457–2471. 2005.
View Article : Google Scholar
|
|
25.
|
Wright CJ, Agboke F, Chen F, La P, Yang G
and Dennery PA: NO inhibits hyperoxia-induced NF-κB activation in
neonatal pulmonary microvascular endothelial cells. Pediatr Res.
68:484–489. 2010.
|
|
26.
|
Blais V and Rivest S: Inhibitory action of
nitric oxide on circulating tumor necrosis factor-induced NF-kappaB
activity and COX-2 transcription in the endothelium of the brain
capillaries. J Neuropathol Exp Neurol. 60:893–905. 2001.
|
|
27.
|
Ho FM, Lin WW, Chen BC, et al: High
glucose-induced apoptosis in human vascular endothelial cells is
mediated through NF-kappaB and c-Jun NH2-terminal kinase
pathway and prevented by PI3K/ Akt/eNOS pathway. Cell Signal.
18:391–399. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Kwan CY, Zhang WB, Deyama T and Nishibe S:
Endothelium-dependent vascular relaxation induced by Eucommia
ulmoides Oliv. bark extract is mediated by NO and EDHF in small
vessels. Naunyn Schmiedebergs Arch Pharmacol. 369:206–211.
2004.PubMed/NCBI
|
|
29.
|
Takemoto M, Sun J, Hiroki J, Shimokawa H
and Liao JK: Rho-kinase mediates hypoxia-induced downregulation of
endothelial nitric oxide synthase. Circulation. 106:57–62. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Steudel W, Scherrer-Crosbie M, Bloch KD,
et al: Sustained pulmonary hypertension and right ventricular
hypertrophy after chronic hypoxia in mice with congenital
deficiency of nitric oxide synthase 3. J Clin Invest.
101:2468–2477. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Fineman JR, Wong J, Morin FC III, Wild LM
and Soifer SJ: Chronic nitric oxide inhibition in utero produces
persistent pulmonary hypertension in newborn lambs. J Clin Invest.
93:2675–2683. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Matsushita H, Morishita R, Nata T, et al:
Hypoxia-induced endothelial apoptosis through nuclear factor-kappaB
(NF-kappaB)-mediated bcl-2 suppression: in vivo evidence of the
importance of NF-kappaB in endothelial cell regulation. Circ Res.
86:974–981. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Chen G, Chen Y, Chen H, et al: The effect
of NF-kappaB pathway on proliferation and apoptosis of human
umbilical vein endothelial cells induced by intermittent high
glucose. Mol Cell Biochem. 347:127–133. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Huang J, Kaminski PM, Edwards JG, et al:
Pyrrolidine dithiocarbamate restores endothelial cell membrane
integrity and attenuates monocrotaline-induced pulmonary artery
hypertension. Am J Physiol Lung Cell Mol Physiol. 294:L1250–L1259.
2008. View Article : Google Scholar
|
|
35.
|
Greco R, Mangione AS, Amantea D, Bagetta
G, Nappi G and Tassorelli C: IkappaB-alpha expression following
transient focal cerebral ischemia is modulated by nitric oxide.
Brain Res. 1372:145–151. 2011. View Article : Google Scholar
|
|
36.
|
Samavati L, Monick MM, Sanlioglu S,
Buettner GR, Oberley LW and Hunninghake GW: Mitochondrial K(ATP)
channel openers activate the ERK kinase by an oxidant-dependent
mechanism. Am J Physiol Cell Physiol. 283:C273–C281. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Harada N, Miura T, Dairaku Y, et al: NO
donor-activated PKC-delta plays a pivotal role in ischemic
myocardial protection through accelerated opening of mitochondrial
K-ATP channels. J Cardiovasc Pharmacol. 44:35–41. 2004. View Article : Google Scholar
|