Introduction
Osteosarcoma (OS) is the most common specific
malignant bone tumor. The five-year survival rate of patients with
OS has increased to 60% with the current combination of modern
surgery and systemic chemotherapy (1). Cisplatin (DDP), which functions in a
manner similar to alkylating agents, is one of the most effective
drugs against OS. However, acquired resistance to DDP by tumor
cells significantly limits its efficacy for the treatment of OS.
Such resistance is closely associated with the recurrence and
metastasis of OS following traditional treatment. The failure of
DDP to induce apoptosis programmed cell death (PCD) type I is
considered one of the major mechanisms underlying resistance to DDP
(2,3). Effective systemic therapeutic
options are therefore required to eliminate the primary lesion,
particularly in drug-resistant OS.
Autophagy is a catabolic process whereby cells
maintain homeostasis by eliminating unnecessary proteins and
damaged organelles (4,5). Autophagy is associated with a number
of physiological processes, including development, differentiation,
neurodegeneration, infection and cancer (6). Previous studies have demonstrated
that autophagy induced in cancer cells by anticancer drugs may
contribute to cancer cell survival in growth-limiting conditions,
such as nutrient depletion, hypoxia, absence of growth factors and
anticancer drug treatments (7–12).
Furthermore, autophagy protects cancer cells from anticancer
drug-induced apoptosis and promotes their survival and recovery
following treatment with chemotherapeutic drugs (13,14).
p62/sequestosome 1 (SQSTM1) is an ubiquitin-binding
protein involved in cell signaling, oxidative stress and autophagy
(15–19). Considering that the induction of
autophagy is accompanied by p62/SQSTM1 protein degradation,
(autophagy usually downregulates the p62 protein level by
degradation, not DNA transcription/tranlation), determining the
intracellular level of p62/SQSTM1 by western blot analysis is used
routinely to measure the autophagic flux in response to
pro-autophagic stimuli. It has been reported that silencing the
expression of the microtubule-associated protein 1 light chain 3
(LC3) protein triggers the intracellular accumulation of
ubiquitinated protein aggregates bound to p62/SQSTM1 and/or
neighbor of BRCA1 gene 1 (NBR1) protein (18,19). Linares et al showed that
the CDK1-dependent phosphorylation of p62/SQSTM1 places a restraint
on tumor transformation, highlighting another function of
p62/SQSTM1, which can act as an oncogene depending on the cellular
circumstances (20).
In this study, we demonstrate that autophagy is
induced by DDP in the Saos-2 cell line, an OS cell that does not
respond to DDP with apoptosis. The induced autophagy protected the
Saos-2 cells from apoptotic cell death induced by DDP. Moreover,
the inhibition of autophagy by chloroquine, an autophagic inhibitor
that blocks lysosomal degradation, accelerated the DDP-induced cell
death of Saos-2 cells. Thus, autophagy appears to protect
drug-resistant OS cells from DDP-induced apoptosis. Moreover, our
data indicate that chloroquine suppresses the autophagic process in
DDP-resistant OS cells by regulating the expression of p62/SQSTM1,
which plays a critical role in the progression and the drug
resistance of OS.
Materials and methods
Cell culture
The established human OS cell lines, MG-63
(CRL-1427™; ATCC, Manassas, VA, USA), U-2OS (HTB-96™; ATCC),
MNNG/HOS (CRL-1547™; ATCC) and Saos-2 (HTB-85; ATCC), were obtained
from the Cell Bank of the Shanghai Institute of Biochemistry and
Cell Biology, Chinese Academy of Sciences (Shanghai, China), where
they were tested and authenticated. All cell lines were grown in
culture medium supplemented with 1% penicillin/streptomycin and 10%
(MG-63, U-2OS and MNNG/HOS) or 15% (Saos-2) (v/v) fetal bovine
serum (Gibco, Grand Island, NY, USA) at 37°C in 5%
CO2.
Reagents and antibodies
DDP (P4394), E-64d (E8640), chloroquine (C6628) and
pepstatin A (P5318) were obtained from Sigma-Aldrich (St. Louis,
MO, USA). The anti-LC3 and anti-p62/SQSTM1 antibodies were
purchased from Sigma-Aldrich, and the antibodies directed against
caspase-3, autophagy protein 5 (ATG5), beclin 1, Barkor/ATG14 and
phosphatidylinositol-3-kinase (PI3K) C3 were purchased from Cell
Signaling Technology (Danvers, MA, USA).
Cell Counting kit-8 (CCK-8) viability
assay
The cells were seeded at 8×103 (MG-63,
HOS and U-2OS) and 1×104 (Saos-2) cells/well in 96-well
plates, with concentrations of 0–50 μM DDP for 48 h and incubated
for an additional 60 min at 37°C in 10% CCK-8 dye (Dojindo, CK04).
Water-soluble tetrazolium salt (WST-8) is reduced by the
dehydrogenases in the cells to create an orange-colored product
(formazan) that is soluble in the cell culture medium. The amount
of the formazan dye generated by the dehydrogenases in the cells is
directly proportional to the number of living cells and is
determined by the absorbance at 450 nm.
Flow cytometry: quantification of
apoptosis
To evaluate the level of apoptosis, the cells
floating in the medium and the adherent cells were collected after
48 h of treatment. The cells were stained using the Annexin V-FITC
Apoptosis Detection kit (Sigma-Aldrich) according to the
manufacturer's instructions. The samples were analyzed using a
FACSCalibur flow cytometer (Becton-Dickinson, North Ryde, New South
Wales, Australia).
Electron microscopy
Untreated and treated cells were fixed in 2.5%
glutaraldehyde at room temperature for 40 min. Cells were
post-fixed in 1% osmium tetroxide at room temperature for 60 min,
dehydrated in a series of increasing concentrations of ethanol
solutions (50, 70, 95 and 100%), and embedded in epoxy resin prior
to sectioning. Representative regions were selected for ultra-thin
sectioning and the samples were examined under an electron
microscope.
Green fluorescent protein (GFP)-LC3
transfection
The GFP-LC3 expression vector was used to visualize
the formation of autophagic vesicles. Cells at 90% confluency were
transiently transfected with GFP-LC3 using Lipofectamine 2000
(Invitrogen, Carlsbad, CA, USA) according to the manufacturer's
instructions. To obtain the highest transfection efficiency and
lowest cytotoxicity, we optimized the ratio of the DNA to
Lipofectamine 2000. Twenty-four hours post-transfection, the cells
were treated with different reagents, fixed with 4%
paraformaldehyde in PBS and placed on slides. The assessment of GFP
fluorescence was carried out using a fluorescence microscope
(Nikon, Tokyo, Japan).
Western blot analysis
The cells were seeded in cell culture flasks or
6-well plates and cultured until they reached approximately 80%
confluency. Fresh medium was added before further processing. Total
cellular protein extracts were prepared by scraping the cells into
Mammalian Protein Extraction Reagent (78503; Thermo Scientific,
Waltham, MA, USA). The protein concentration was measured using a
BCA protein assay (Thermo Scientific Pierce). The proteins were
separated by SDS-PAGE 10–15% and electrotransferred onto either
nitrocellulose or polyvinylidene fluoride (PVDF) membranes
(Millipore, Billerica, MA, USA). The membranes were blocked with 5%
bovine serum albumin (BSA) in Tris-buffered saline containing 0.1%
Tween-20 (TBST, pH 7.6) for 90 min at room temperature. All primary
antibodies were incubated overnight at 4°C. The membranes were
incubated in secondary antibodies diluted in 5% BSA in TBST for 90
min at room temperature with mild agitation. The membranes were
washed three times between each of the incubations. The labeled
proteins were detected by enhanced chemiluminescence (ECL kit;
Bio-Rad, Hercules, CA, USA).
Statistical analysis
The data are expressed as the means ± SD. The
statistical significance of the differences was determined using a
Student's two-tailed t-test for two groups and one-way ANOVA for
multiple groups. P-values <0.05 were considered to indicate
statistically significant differences. All data were analyzed using
SPSS 16.0 software.
Results
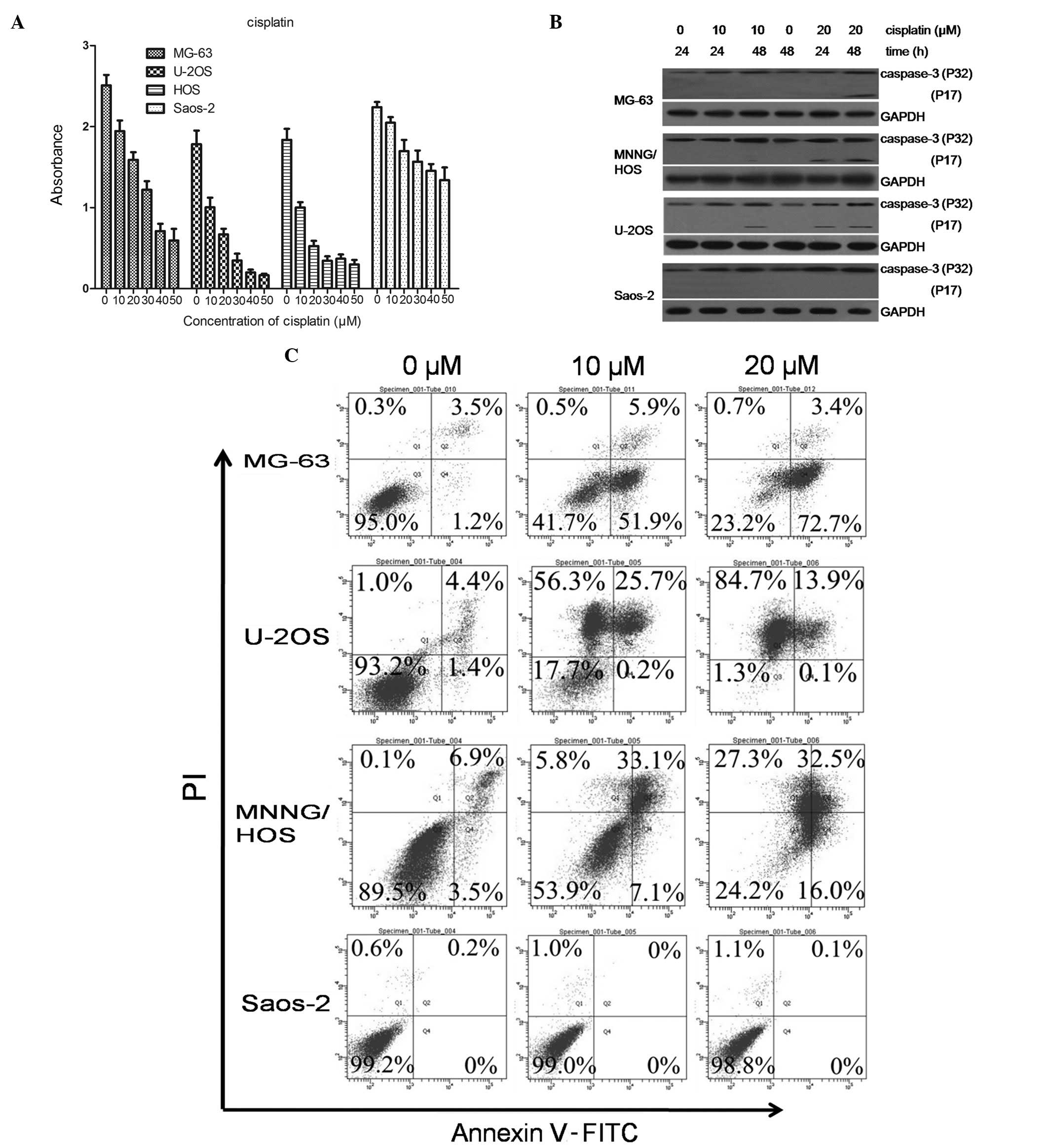
DDP-induced cell death in OS cells
We evaluated a group of four OS cell lines, MG-63,
U-2OS, MNNG/HOS and Saos-2, for their sensitivity to the
chemotherapeutic drug, DDP. DDP (10 μM) inhibited cell
proliferation in the U-2OS and MNNG/HOS cells. The MG-63 cells were
marginally more resistant to DDP treatment compared to the U-2OS
and MNNG/HOS cells, and the Saos-2 cells were completely resistant
to DDP treatment at 10 μM or even higher concentrations (Fig. 1A).
We then examined the typical markers of apoptotic
cell death in the four cell lines. Th expression of cleaved
caspase-3 was assessed following treatment with DDP (Fig. 1B). The expression of cleaved
caspase-3 was significantly increased in the U-2OS, MNNG/HOS and
MG-63 cells, whereas no induction of cleaved caspase-3 expression
was observed in the Saos-2 cells. To quantitatively evaluate
apoptosis in the four cell lines, we exposed the cells to 0, 10 or
20 μM DDP for 48 h, and the apoptosis rate was determined using the
Annexin V-PI (AV-PI) assay. Fig.
1C shows the flow cytometric analyses representative of three
independent experiments. Both the drug-sensitive (U-2OS and
MNNG/HOS) cells and the relatively resistant MG-63 cells
demonstrated necrosis and apoptosis (76.1% apoptosis rate and 0.7%
necrosis rate for MG-63, 14% apoptosis rate and 84.7% necrosis rate
for U-2OS, 48.5% apoptosis rate and 27.3% necrosis rate for
MNNG/HOS) in response to DDP (20 μM). By contrast, the
significantly drug-resistant (Saos-2) cells had a very low rate of
apoptosis (0.1%).
DDP induces authentic autophagy in the
Saos-2 cells Comparison of the morphological features of four
DDP-treated OS cells observed under an electron microscope
The U-2OS, MG-63 and MNNG/HOS cells treated with DDP
(40 μM) for 48 h showed typical apoptotic morphology, including
cell shrinkage and chromatin marginalization with an intact but
blebbed plasma membrane and late apoptotic phenomena, such as
nuclear debris and apoptotic body formation (Fig. 2A). By contrast, the DDP-treated
Saos-2 cells retained an intact nuclear membrane containing a
distinct nucleolus, with areas of more electron-dense
heterochromatin. In addition, higher magnification images revealed
the presence of numerous cytoplasmic vacuoles resembling nascent
autophagosomes, many of which appeared to surround cytoplasmic
material and components, such as the mitochondria (Fig. 2B).
DDP induces the formation and degradation
of autophagosomes in Saos-2 cells
LC3, an autophagosomal membrane protein, is widely
used as a cellular marker to assess the quantity of autophagosomes
in cells (21). LC3 with an
N-terminus GFP has been used to monitor autophagy through direct
fluorescence microscopy. Diffuse GFP fluorescence was observed in
the U-2OS, MG-63 and MNNG/HOS and Saos-2 cells expressing
GFP-tagged LC3 using a laser scanning confocal fluorescence
microscope. Moreover, the number of GFP-LC3 puncta in the Saos-2
cells significantly increased following exposure to DDP (48 h)
(Fig. 3A). These results suggest
that DDP induces autophagy in the Saos-2 cells.
The formation of a lipidation form of LC3, termed
LC3-II is induced by autophagy. Thus, it is a reliable biochemical
marker for autophagy. We therefore investigated the conversion of
LC3 form I (LC3-I, 18 kDa) to form II (LC3-II, 16 kDa) by western
blot analysis. The results revealed that the U-2OS, MG-63 and
MNNG/HOS cells did not convert LC3-I to LC3-II following exposure
to DDP. By contrast, the levels of LC3-I and LC3-II were
downregulated in the Saos-2 cells that were treated with DDP for 48
h (Fig. 3B). This is may be due
to the rapid lysosomal degradation of LC3-II during autophagy.
Thus, we believe that DDP indeed induces autophagy in the Saos-2
cells.
To elucidate the molecular mechanism by which DDP
induces autophagy in the Saos-2 cells, we co-treated the cells with
different autophagic inhibitors. As shown in Fig. 3Ci, GFP-LC3 puncta were not
observed when the cells were treated with the lysosomal protease
inhibitor, chloroquine (6 μM, added 2 h prior to exposure to DDP);
however, the degradation of autophagosomes was not blocked by the
other inhibitors of lysosomal proteases, E64d and pepstatin A. The
average number of GFP-LC3-positive puncta in ten typical (x63
magnifications) cells was counted to measure the statistical
differences. Histograms revealed that only chloroquine affected the
degradation of the autophagosomes (Fig. 3Di). This is consistent with the
fluorescence microscopy data. The results from western blot
analysis demonstrated that LC3-II was significantly upregulated in
the Saos-2 cells treated with a combination of DDP and chloroquine,
compared to the Saos-2 cells treated with DDP alone (Fig. 3E). However, the accumulation of
LC3-II and the inhibition of lysosomal proteases was not observed
in the MG-63 cells, which are mildly resistant to DDP-induced
apoptosis (Fig. 3Cii and
Dii).
Taken together, these results provided strong
support to the conclusion that while DDP induces apoptosis in the
U-2OS, MG-63, MNNG/HOS cells it causes autophagy in the Saos-2
cells. Furthermore, autophagy protected the Saos-2 cells from
DDP-induced cell death.
Chloroquine blocks the DDP-induced
autophagy in Saos-2 cells by regulating p62/SQSTM1
The protein levels of autophagic substrates can be
used to monitor the autophagic flux. A previous study revealed that
several specific substrates are preferentially degraded by
autophagy, among which the most extensively studied autophagic
substrate, is p62/SQSTM1 (22).
In this study, we examined the expression of p62/SQSTM1 in the
Saos-2 and MG-63 cells treated with 0, 10 and 20 μM DDP for 48 h.
The downregulation of p62/SQSTM1 was observed in the Saos-2 cells
48 h following exposure to DDP (Fig.
4A). To examine such a regulation in further detail, we
examined the expression of p62/SQSTM1 at various time points over a
period of 24 h in the Saos-2 and MG-63 cells treated with 20 μM
DDP. In the Saos-2 cells, the expression level of p62/SQSTM1
decreased after 1 h of treatment with DDP, followed by a gradual
recovery (Fig. 4B). These results
suggest that the total cellular expression levels of p62/SQSTM1
correlate with the autophagic activity and that p62/SQSTM1 plays a
potential role in the response to DDP in the Saos-2 cells. A
previous study revealed that the inhibition of autophagy in HeLa
cells resulted in the accumulation of p62/SQSTM1 (22). As shown in Fig. 4C, the downregulation of p62/SQSTM1
expression was observed when autophagy was inhibited by the
lysosomal protease inhibitor, chloroquine (6 μM, added 2 h prior to
exposure to DDP); however, the formation of autophagosomes was not
blocked by E64d or pepstatin A, two other inhibitors of lysosomal
proteases. Accordingly, we found that DDP in combination with
chloroquine inhibited cell proliferation in a dose-dependent manner
(Fig. 4D). These data suggest
that chloroquine inhibits the autophagic process in DDP-resistant
OS cells by regulating the expression or degradation of
p62/SQSTM1.
Discussion
In recent decades, although the survival rate of
patients with OS has increased as a result of rapid advancements in
comprehensive therapy, particularly neoadjuvant chemotherapy, the
cytotoxic effects of drugs on OS cells are reduced by acquired
chemoresistance. Therefore, the specific mechanisms of drug
resistance and the molecular targets involved must be explored to
overcome the resistance to cytotoxic drugs. Autophagy may help
cancer cells survive in growth-limiting conditions, such as the
presence of anticancer drugs. In this study, we demonstrate that
DDP-sensitive cells underwent authentic apoptosis, with the cells
exhibiting caspase-dependent death. Of note, only a small number of
Saos-2 cells underwent apoptosis following treatment with DDP.
Furthermore, the results revealed that DDP induced autophagy in a
dose- and time-dependent manner in this DDP-resistant cell line,
which may be the survival factor facilitating their development of
acquired resistance.
Autophagy is a catabolic process for the
autophagosomic-lysosomal degradation of bulk cytoplasmic content.
The autophagic flux can be measured by measuring the LC3-II
turnover in the presence or absence of lysosomal degradation, which
can be assessed by western blot analysis (20). Lysosomal degradation can be
prevented by certain protease inhibitors (e.g., chloroquine,
pepstatin A and E64d) (23). In
our study, despite the salient attenuation of LC3-I, the conversion
of endogenous LC3-I to LC3-II in the DDP-treated Saos-2 cells was
insignificant. Therefore, we examined the turnover of LC3-II in the
Saos-2 cells treated with DDP in combination with different
lysosomal inhibitors (chloroquine or pepstatin A and E64d) by
western blot analysis and laser confocal fluorescence microscopy.
The results revealed that the DDP-induced aggregation of LC3-II was
significantly reduced in the Saos-2 cells treated with chloroquine
and DDP but not when they were treated with chloroquine plus
pepstatin A and E64d. These results were confirmed by the
fluorescent data, which demonstrated the accumulation of GFP-LC3
puncta in the Saos-2 cells treated with DDP in combination with
lysosomal protease inhibitors. In addition, the levels of other
autophagic substrates may be used to monitor the autophagic flux.
The adaptor protein, p62/SQSTM1, is required for the formation of
ubiquitinated protein aggregates in vitro. p62/SQSTM1 is
selectively incorporated into autophagosomes through its direct
binding with LC3. Therefore, the total cellular expression levels
of p62/SQSTM1 inversely correlate with autophagic activity. In our
study, our results revealed that the expression level of p62/SQSTM1
was upregulated following treatment with DDP for 48 h. These
results suggest that the analysis of p62/SQSTM1 expression may be
useful in assessing the early induction of autophagy and may also
be a marker of autophagy. More importantly, p62/SQSTM1 plays a
significant role in the resistance of OS cells to DDP.
Chloroquine has been used for the treatment of
various diseases, such as malaria and rheumatoid arthritis for
several decades (23). The
application of chloroquine as an adjuvant anticancer drug has
previously been reported as it plays a key role in halting the DNA
repair process. In addition, treatment with chloroquine leads to an
apparent increase in the formation of autophagosomes, possibly by
blocking the fusion of lysosomes (24). For this reason, in this study, we
investigated the potential cytotoxic effects of three lysosomal
protease inhibitors (chloroquine, E64d and pepstatin A).
Furthermore, we assessed the contribution of DDP-induced autophagy
to the survival of Saos-2 cells, using the lysosomotropic agent
chloroquine, which prevents lysosomal degradation. The marked
increase in cell death observed following co-treatment with DDP and
chloroquine suggested that the addition of a late phase autophagic
inhibitor to DDP reduced the chemoresistance of the Saos-2 cells.
Notably, the addition of chloroquine diminished the DDP-induced
downregulation of p62/SQSTM1 expression in the Saos-2 cells,
suggesting that chloroquine blocked the DDP-induced autophagy in
the drug-resistant OS cells by regulating the expression of
p62/SQSTM1 and LC3. The combination of an autophagic inhibitor with
agents that induce apoptosis in drug-resistant cancer cells as a
survival response has recently been proposed as a novel targeted
therapy for cancer (25,26). Thus, chloroquine appears to be a
promising candidate for the inhibition of autophagy in OS cells,
which has long been used as an anti-malarial and anti-rheumatic
drug.
In conclusion, in this study, we demonstrate that
autophagy is a protective mechanism induced in DDP-resistant human
OS cells in vitro. In addition, chloroquine dramatically
suppressed the autophagic process that led to apoptotic cell death
in the DDP-resistant OS cells by regulating the expression of LC3
and p62/SQSTM1. Our data suggest a potential clinical therapy
targeting autophagy with chloroquine or monoclonal antibodies for
the treatment of drug-resistant OS.
Acknowledgements
This study was supported in part by a grant
(2012C24008) from the Science Technology Department of Zhejiang
Province. We thank Qing Zhong (University of California at
Berkeley) for providing the GFP-LC3 plasmid.
References
|
1
|
Bacci G, Ferrari S, Bertoni F, et al:
Long-term outcome for patients with nonmetastatic osteosarcoma of
the extremity treated at the istituto ortopedico rizzoli according
to the istituto ortopedico rizzoli/osteosarcoma-2 protocol: an
updated report. J Clin Oncol. 18:4016–4027. 2000.
|
|
2
|
Brozovic A and Osmak M: Activation of
mitogen-activated protein kinases by cisplatin and their role in
cisplatin-resistance. Cancer Lett. 251:1–16. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gorlick R, Anderson P, Andrulis I, et al:
Biology of childhood osteogenic sarcoma and potential targets for
therapeutic development: meeting summary. Clin Cancer Res.
9:5442–5453. 2003.PubMed/NCBI
|
|
4
|
Reggiori F and Klionsky DJ: Autophagy in
the eukaryotic cell. Eukaryot Cell. 1:11–21. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Codogno P and Meijer AJ: Autophagy and
signaling: their role in cell survival and cell death. Cell Death
Differ. 12(Suppl 2): 1509–1518. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Levine B and Yuan J: Autophagy in cell
death: an innocent convict? J Clin Invest. 115:2679–2688. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kondo Y, Kanzawa T, Sawaya R and Kondo S:
The role of autophagy in cancer development and response to
therapy. Nat Rev Cancer. 5:726–734. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Krysko DV and Vandenabeele P: From
regulation of dying cell engulfment to development of anti-cancer
therapy. Cell Death Differ. 15:29–38. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Degenhardt K, Mathew R, Beaudoin B, et al:
Autophagy promotes tumor cell survival and restricts necrosis,
inflammation, and tumorigenesis. Cancer Cell. 10:51–64. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Amaravadi RK, Yu D, Lum JJ, et al:
Autophagy inhibition enhances therapy-induced apoptosis in a
Myc-induced model of lymphoma. J Clin Invest. 117:326–336. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Rosenfeldt MT and Ryan KM: The role of
autophagy in tumour development and cancer therapy. Expert Rev Mol
Med. 11:e362009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jin S and White E: Tumor suppression by
autophagy through the management of metabolic stress. Autophagy.
4:563–566. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
O'Donovan TR, O'Sullivan GC and McKenna
SL: Induction of autophagy by drug-resistant esophageal cancer
cells promotes their survival and recovery following treatment with
chemotherapeutics. Autophagy. 7:509–524. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sun WL, Chen J, Wang YP and Zheng H:
Autophagy protects breast cancer cells from epirubicin-induced
apoptosis and facilitates epirubicin-resistance development.
Autophagy. 7:1035–1044. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Seibenhener ML, Geetha T and Wooten MW:
Sequestosome 1/p62 - more than just a scaffold. FEBS Lett.
581:175–179. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Komatsu M, Kurokawa H, Waguri S, et al:
The selective autophagy substrate p62 activates the stress
responsive transcription factor Nrf2 through inactivation of Keap1.
Nat Cell Biol. 12:213–223. 2010.PubMed/NCBI
|
|
17
|
Bjorkoy G, Lamark T and Johansen T:
p62/SQSTM1: a missing link between protein aggregates and the
autophagy machinery. Autophagy. 2:138–139. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kirkin V, McEwan DG, Novak I and Dikic I:
A role for ubiquitin in selective autophagy. Mol Cell. 34:259–269.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shvets E, Fass E, Scherz-Shouval R and
Elazar Z: The N-terminus and Phe52 residue of LC3 recruit
p62/SQSTM1 into autophagosomes. J Cell Sci. 121:2685–2695. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Linares JF, Amanchy R, Greis K, Diaz-Meco
MT and Moscat J: Phosphorylation of p62 by cdk1 controls the timely
transit of cells through mitosis and tumor cell proliferation. Mol
Cell Biol. 31:105–117. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kabeya Y, Mizushima N, Ueno T, et al: LC3,
a mammalian homologue of yeast Apg8p, is localized in autophagosome
membranes after processing. EMBO J. 19:5720–5728. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bjorkoy G, Lamark T, Brech A, et al:
p62/SQSTM1 forms protein aggregates degraded by autophagy and has a
protective effect on huntingtin-induced cell death. J Cell Biol.
171:603–614. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Solomon VR and Lee H: Chloroquine and its
analogs: a new promise of an old drug for effective and safe cancer
therapies. Eur J Pharmacol. 625:220–233. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tanida I, Minematsu-Ikeguchi N, Ueno T and
Kominami E: Lysosomal turnover, but not a cellular level, of
endogenous LC3 is a marker for autophagy. Autophagy. 1:84–91. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kaini RR and Hu CA: Synergistic killing
effect of chloroquine and androgen deprivation in LNCaP cells.
Biochem Biophys Res Commun. 425:150–156. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sasaki K, Tsuno NH, Sunami E, et al:
Chloroquine potentiates the anti-cancer effect of 5-fluorouracil on
colon cancer cells. BMC Cancer. 10:3702010. View Article : Google Scholar : PubMed/NCBI
|