Introduction
The early repolarization pattern consisting of a
J-point elevation, notching or slurring of the terminal portion of
the R wave (J wave) and tall/symmetric T wave is a common finding
on the electrocardiography (ECG) in young healthy men or athletes
and is considered a ‘benign’ ECG manifestation for a long period of
time (1–3). However, recent evidence has
demonstrated that early repolarization may be associated with an
increased risk for ventricular fibrillation (VF), depending on the
locations and magnitude of the J wave and ST-segment elevation
(4,5). Thus, Antzelevitch and Yan (1) proposed a new conceptual framework,
inherited J wave syndromes, to describe the arrhythmic phenotypes.
Inherited J wave syndromes, as a continuous spectrum of arrhythmic
phenotypes, has been divided into four different subtypes in terms
of anatomical locations of J wave and ST-segment elevation and
clinical consequences, including early repolarization in the
lateral precordial leads [early repolarization syndrome (ERS) type
1], in the inferior or inferolateral leads (ERS type 2), globally
in the inferior, lateral and right precordial leads (ERS type 3)
and in the right precordial leads [Brugada syndrome (BrS)]
(1). However, the genetic basis
of the three types of ERS has not been well defined. To date,
‘loss-of-function’ mutations in the CACNA1C, CACNB2
and CACNA2D1 genes and ‘gain-of-function’ mutations in the
KCNJ8 gene have been identified in patients with ERS
(6–8).
In the present study, we described an unreported
missense mutation of c.4297 G>C in the SCN5A gene, which
encoded the α subunit of the cardiac voltage-gated sodium channel
Nav1.5, leading to prominent J waves and ST-segment elevation in
the inferior, lateral and precordial leads accompanied with VF in a
female proband. We identified an unpublished synonymous single
nucleotide polymorphism (SNP) of T5457C in the SCN5A gene in
the proband, her mother and sister. We characterized the functional
consequences of the mutation as well as the interaction between the
mutation and the SNP.
Materials and methods
Clinical examination
The proband and her first-degree relatives underwent
clinical evaluation, including medical history, physical
examination and 12-lead ECG and echocardiographic examinations
(Figs. 1 and 2). The study conformed to the principles
outlined in the Declaration of Helsinki. The institutional ethics
committee approved all the research protocols and written informed
consents were obtained from all participants.
Genotyping
Genomic DNA was isolated from peripheral blood
leukocytes using a TIANamp Blood DNA isolation kit (Tiangen,
Beijing, China) according to the manufacturer’s instructions. All
exons of the SCN5A, KCND3, CACNA1C,
CACNB2, KCNE3, SCN1B and KCNJ8 genes
were amplified by polymerase chain reaction (PCR) and were analyzed
by direct sequencing. Whenever the variant was discovered in the
proband or her family members, it would be confirmed in 500
unrelated healthy Chinese individuals (1,000 reference alleles) to
determine the distinction between mutation and polymorphism.
Mutagenesis and expression studies
The technique for site-directed mutagenesis and
wild-type (WT)-SCN5A plasmid were prepared in our laboratory
(9). Three mutations based on the
genotypes of the patient and her family (c.4297 G>C -SCN5A,
T5457C-SCN5A and c.4297 G>C-T5457C-SCN5A) were created on the
WT-SCN5A background (GenBank ID: NM198056). All mutations were
verified by sequencing and then subcloned back into the pcDNA3.1
vector. The heterologous expression was performed as previously
described (9). Briefly, the human
embryonic kidney (HEK) 293 cells were transiently transfected with
0.6 μg cDNA (detailed composition is shown in Table I) using an Effectene Transfection
Reagent (Qiagen, Hilden, Germany) according to the manufacturer’s
protocol. Green fluorescent protein gene (0.2 μg) was cotransfected
as an indicator. The transfected cells were cultured for 36 h
before sodium current (INa) was recorded. More
than 3 independent experiments were conducted to confirm the
reproducibility of the results.
 | Table IAverage values of activation,
inactivation and recovery from inactivation parameters. |
Table I
Average values of activation,
inactivation and recovery from inactivation parameters.
| | Steady-state
activation | Steady-state
inactivation | Recovery from
inactivation |
|---|
| |
|
|
|
|---|
| Channels | N | V1/2,
mV | k | V1/2,
mV | k | τf,
msec | τs,
msec | Af |
|---|
| WT-SCN5A (0.6
μg) | 8 | −50.72±1.46 | 3.61±0.32 | −88.61±0.78 | 5.28±0.14 | 24.25±4.26 | 189.94±14.56 | 0.70±0.01 |
| G4297C-SCN5A (0.6
μg) | 9 | −39.07±1.80a | 6.18±0.36a | −91.39±1.95 | 4.79±0.47 | 66.01±4.94a | 257.99±8.84a | 0.54±0.01a |
| T5457C-SCN5A (0.6
μg) | 8 | −47.26±2.34 | 4.50±0.54 | −90.91±1.00 | 5.35±0.17 | 29.38±3.44 | 248.93±6.95a | 0.50±0.02a |
| G4297C-T5457C-SCN5A
(0.6 μg) | 8 | −40.78±2.19a | 5.29±0.35a | −85.71±1.96 | 4.78±0.31 | 56.46±2.48a | 244.10±9.11a | 0.52±0.01a |
| G4297C-SCN5A +
T5457C-SCN5A (0.3 + 0.3 μg) | 7 | −39.10±2.69a | 5.46±0.42a | −88.64±0.86 | 5.03±0.25 | 46.43±4.36a |
232.85±10.55a | 0.50±0.01a |
| WT-SCN5A +
G4297C-SCN5A (0.3 + 0.3 μg) | 5 | −46.01±2.57 | 4.91±0.68 | −87.56±0.98 | 5.55±0.16 | 25.92±4.20 | 204.75±9.81 | 0.69±0.01 |
| WT-SCN5A +
T5457C-SCN5A (0.3 + 0.3 μg) | 5 | −47.87±3.13 | 3.99±1.80 | −87.97±1.99 | 5.36±0.27 | 27.43±3.65 |
258.76±12.74a | 0.56±0.02a |
| WT-SCN5A +
G4297C-T5457C-SCN5A (0.3 + 0.3 μg) | 9 | −42.60±1.77a | 5.31±0.44a | −85.21±1.53 | 5.09±0.27 | 47.05±6.09a |
236.03±10.13a | 0.49±0.02a |
| T5457C-SCN5A +
G4297C-T5457C-SCN5A (0.3 + 0.3 μg) | 5 | −43.72±2.51a | 4.89±0.43a | −88.67±2.94 | 5.66±0.30 | 51.47±7.14a |
252.87±13.82a | 0.51±0.01a |
Patch-clamping
INa was measured using whole-cell
configuration of the patch-clamp technique as previously described
(10) with Axonpatch 700B
amplifiers (Axon Instruments, Foster City, CA, USA). The resistance
of pipettes in the bath solution ranged from 1.5 to 2 MΩ. For
whole-cell recording, the internal pipette solution contained (in
mmol/l) CsF 120, CsCl 20, ethylene glycol tetraacetic acid 2.0,
MgCl2 1.0 and HEPES 5 (pH 7.4). The bath solution
contained (in mmol/l) NaCl 30, CsCl 110, CaCl2 1.8,
MgCl2 1.0 and HEPES 10 (pH 7.4). No leak subtraction was
applied during recording. All measurements were obtained at room
temperature (20–22°C).
RNA isolation and quantitative real-time
PCR
Total RNA was extracted from transfected HEK293
cells using TRIzol reagent (Invitrogen Life Technologies, Carlsbad,
CA, USA) according to the manufacturer’s instructions. The
concentration of RNA was determined by the UV Visible
spectrophotometer and 1 μg total RNA was subsequently
reverse-transcribed to cDNA using Moloney murine leukemia virus
reverse transcriptase (Promega Corporation, Madison, WI, USA).
Quantitative real-time PCR (11)
was performed on an ABI 7300 real-time PCR instrument (Applied
Biosystems, Foster City, CA, USA) with the GAPDH gene, whose
expression is very stable in HEK293 cells, as an internal control
for data normalization. The primers were as follows: forward,
5′-GAGATGCTGCAGGTCGGAAAC-3′ and reverse,
5′-GATGACGATGATGCTGTCGAAGA-3′. The PCR cycles consisted of
denaturation at 95°C for 15 sec, annealing and extension at 60°C
for 1 min for 40 cycles. Each sample was analyzed 3 times. The
2−ΔΔCt method was used to analyze the relative
expression of protein.
Immunocytochemistry
Immunocytochemical experiments were performed as
previously described (12).
Briefly, cells were fixed, blocked and incubated with mouse
monoclonal to Nav1.5 IgM primary antibody (1:40 dilution) (Abcam,
Cambridge, UK) overnight at 4°C. The cells were then incubated in
anti-mouse FITC-conjugated donkey secondary antibody (1:500
dilution) (Jackson ImmunoResearch, West Grove, PA, USA) for 1 h at
room temperature in the dark. Confocal images were obtained using a
confocal laser scanning microscope (Leica TCS SP2; Leica, Wetzlar,
Germany) and were analyzed with NIH image software (Image J).
Statistical analysis
Data are represented as the means ± standard error
of the mean (SEM). Student’s t-test analysis was performed for the
comparison of two means, while one-way ANOVA was performed for
comparisons of multiple means using statistical software SPSS 13.0.
A value of P<0.05 was considered to indicate a statistically
significant difference.
Results
Clinical manifestation and
genotyping
The proband, a 19-year-old female was admitted to
the Fu Wai Hospital (Beijing, China) due to a history of recurrent
syncope at rest (a total of 4 times since the age of 16). The ECG
that was obtained at the emergency department exhibited prominent J
waves and ST-segment elevation in leads I, II, III, aVF and
V2–V6 (Fig.
2B) before VF occurred (Fig.
2C). Subsequently, the VF episode was terminated by
cardioversion. There was no difference in clinical state that may
explain the marked J waves and ST-segment elevation in the
documented ECG. Myocardial infarction was excluded by cardiac
enzyme tests and coronary angiography. The routine echocardiography
demonstrated a structural and functional normal heart. The baseline
ECG was within normal limits with the exception of tiny, hump-like
J waves in the inferior leads and a borderline prolongation of the
PR interval (200 msec) (Fig. 2A).
The amplitude of ST-segment elevation fluctuated from day to day.
Programmed electrical stimulation induced VF that was converted to
sinus rhythm by cardioversion. Since the sodium channel blockers,
such as ajmaline, flecainide and procainamide, were not available
in China, drug challenge was not performed. According to the
clinical manifestations, ERS type 3 was diagnosed and an
implantable cardioverter defibrillator was implanted with the
pacing rate programmed to 60 bpm. After amiodarone (200 mg/day) and
metoprolol (50 mg/day) administration, the proband remained
symptom-free for a period of 18 months. However, in the nineteenth
month, the patient was shocked due to a VF episode with a heart
rate >260 bpm. All family members lived asymptomatic and there
was no family history of sudden death.
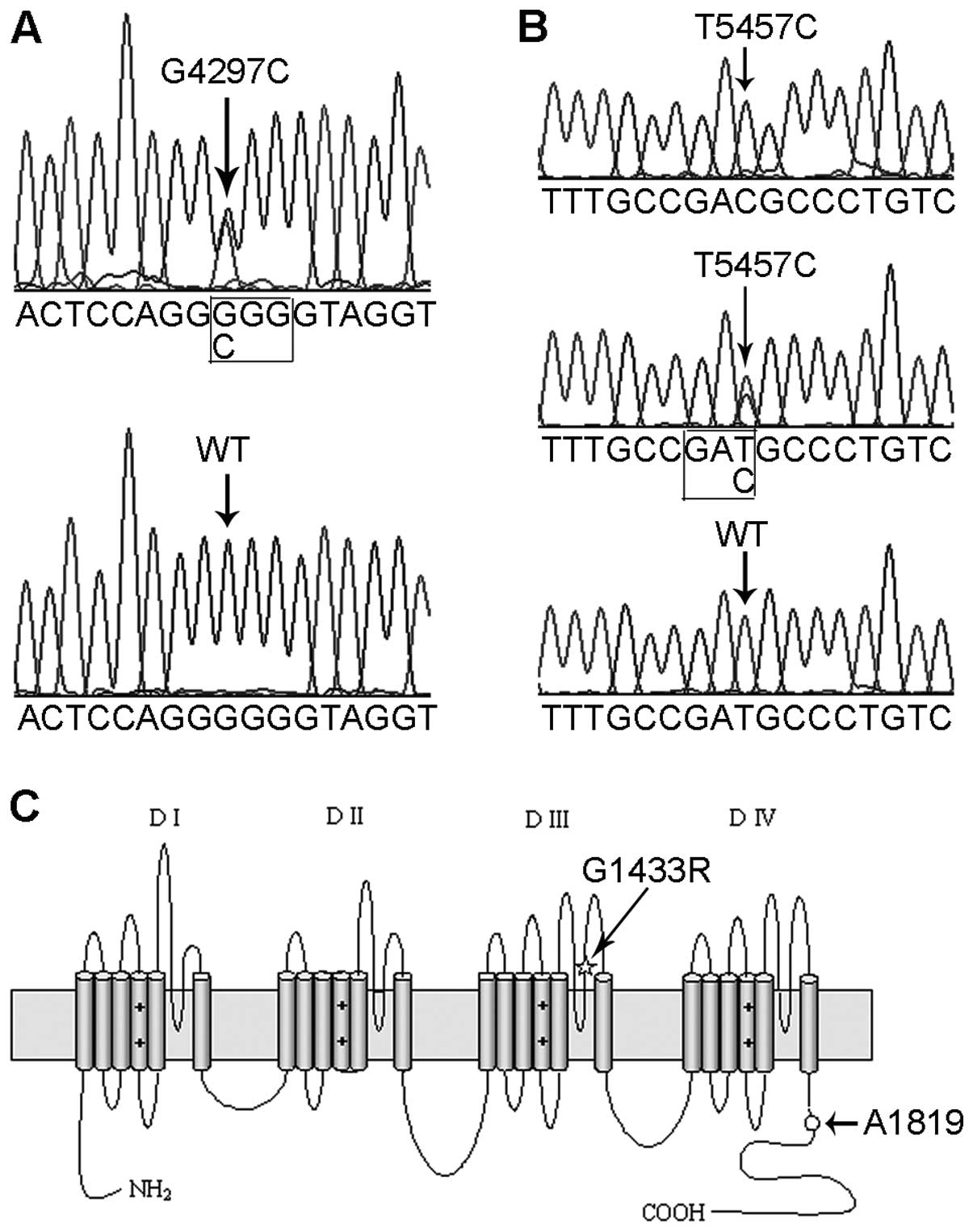
A heterozygous missense mutation of c.4297 G>C,
located in exon 24 of the SCN5A gene, was identified in the
proband using direct DNA sequencing (Fig. 3A). The mutation led to a
substitution of an arginine for a glycine at position 1433 that was
predicted to be in the extracellular loop between the pore region
and the S6 transmembrane segment in the domain III of the α subunit
of the sodium channels (Fig. 3C).
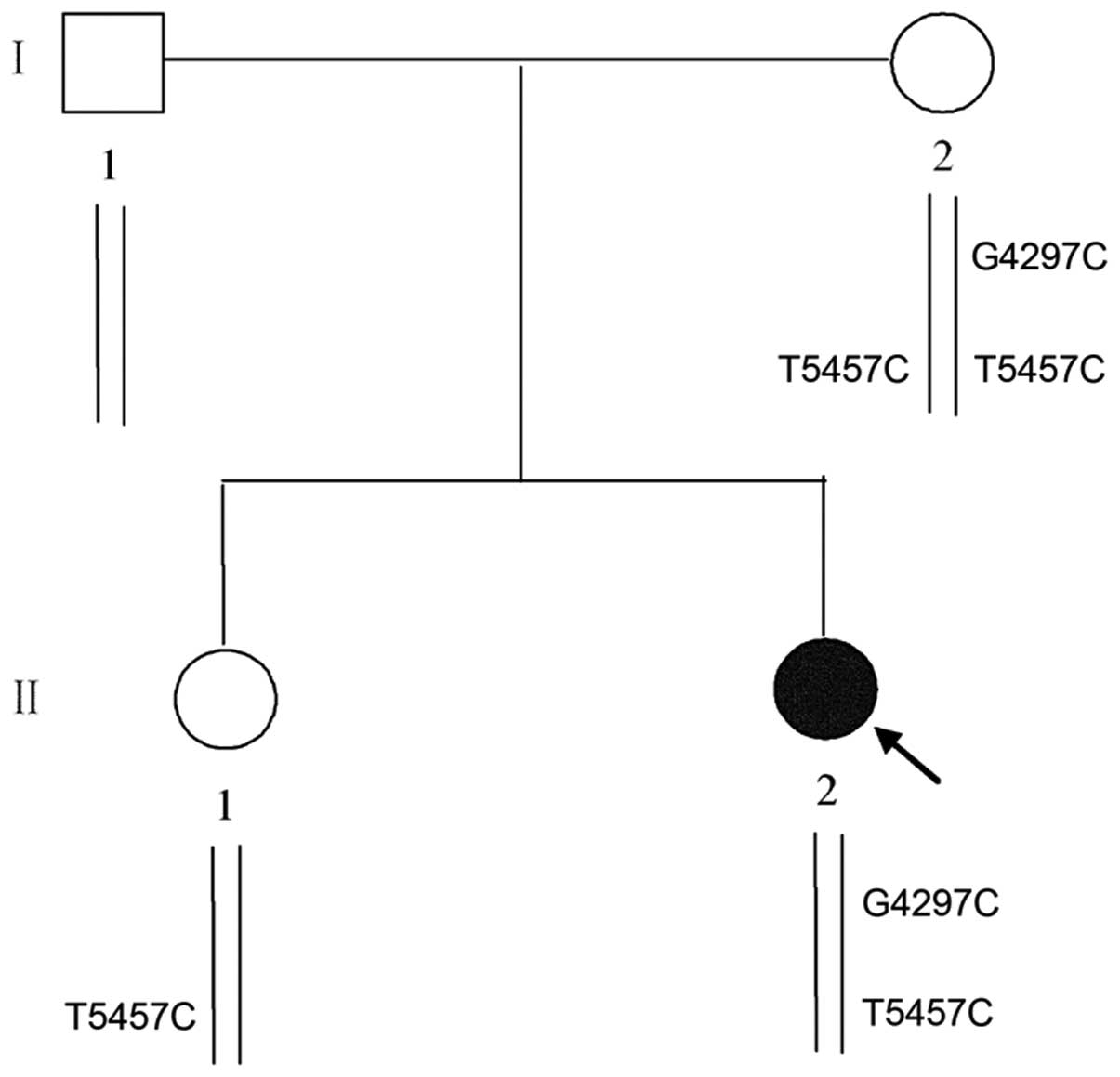
The c.4297 G>C mutation was also detected in her mother but not
in 500 unrelated healthy subjects. In addition, a heterozygous SNP,
T5457C, which is located in exon 28 of the SCN5A gene, was
discovered in the proband; however, the T5457C polymorphism did not
cause a change in the protein sequence (Fig. 3B and C). Genetic analysis revealed
that her mother carried a homozygous T5457C polymorphism and her
sister carried a heterozygous T5457C polymorphism, while her
farther carried no such variants (Fig. 1). Since the substitution of C for
T occurred at a frequency of 42.67% within a population including
500 unrelated healthy subjects, we confirmed that the T5457C
variant was a polymorphism. Moreover, no other mutations or
variants were discovered in the screened genes in the proband and
the family.
Electrophysiology of G4297C and T5457C
channels
INa was recorded from the HEK293
cells transiently transfected with the WT-SCN5A, G4297C-SCN5A or
T5457C-SCN5A plasmid using the whole-cell configuration of the
patch-clamp technique. The G4297C mutation dramatically reduced
INa density (−107.65±14.70 pA/pF in WT, n=8 vs.
−15.75±3.72 pA/pF in the mutant, n=12, P<0.05) (Fig. 4A and C) and shifted the
steady-state activation curve to a more positive potential compared
with the WT channels (P<0.05) (Fig. 5C and Table I). By contrast, the mutation had
no effect on the steady-state inactivation (Fig. 5D and Table I), although it inhibited the
recovery from inactivation (P<0.05) (Fig. 5B and Table I).
The T5457C variant was a relative benign
polymorphism since it did not change the current density
(−115.47±23.25 pA/pF, n=8, P>0.05) (Fig. 4B), steady-state activation and
steady-state inactivation of the sodium channels, although it
prolonged the recovery from inactivation compared with the WT
channels (P<0.05) (Table
I).
Rescue effect of the T5457C polymorphism
on G4297C mutant channels
Furthermore, we characterized the interaction
between the G4297C mutation and the T5457C polymorphism. Inserting
the T5457C polymorphism into the G4297C mutant construct
(G4297C-T5457C-SCN5A) partially restored the INa
density (−44.12±10.45 pA/pF, n=8) (P<0.05, Fig. 4D) but without having an effect on
mutant channel kinetics compared with the G4297C channels (Table I). However, coexpression of the
T5457C polymorphism and G4297C mutation on separate constructs had
no significant effect on the INa density of
mutant channels, since the similar current densities were recorded
from cells coexpressing G4297C-SCN5A and WT or T5457C-SCN5A
(−55.66±18.01 pA/pF, n=7 vs. −61.02±17.80 pA/pF, n=5, P>0.05)
(Fig. 4E and F). Additionally, we
expressed the cDNA constructs to simulate the distribution patterns
of G4297C and T5457C in the proband and her family. Coexpression of
G4297C-T5457C-SCN5A with WT-SCN5A (as the nature of the proband) or
G4297C-T5457C-SCN5A with T5457C-SCN5A (as the nature of her mother)
increased the INa up to 65 or 70% in the WT
channels. The peak current densities are plotted in Fig. 5A for all combinations of WT-SCN5A,
G4297C-SCN5A, T5457C-SCN5A and G4297C-T5457C-SCN5A constructs.
Transcription of WT and mutant cDNAs
Quantitative real-time PCR was used to detect the WT
and mutant mRNA levels. The mRNA of SCN5A was not detected in the
non-transfected cells. There was no significant difference in mRNA
levels between cells transfected with WT-SCN5A and the G4297C
mutant (P>0.05). The mRNA level of G4297C-T5457C-SCN5A was
increased by 6-fold compared to that of WT-SCN5A (P<0.05).
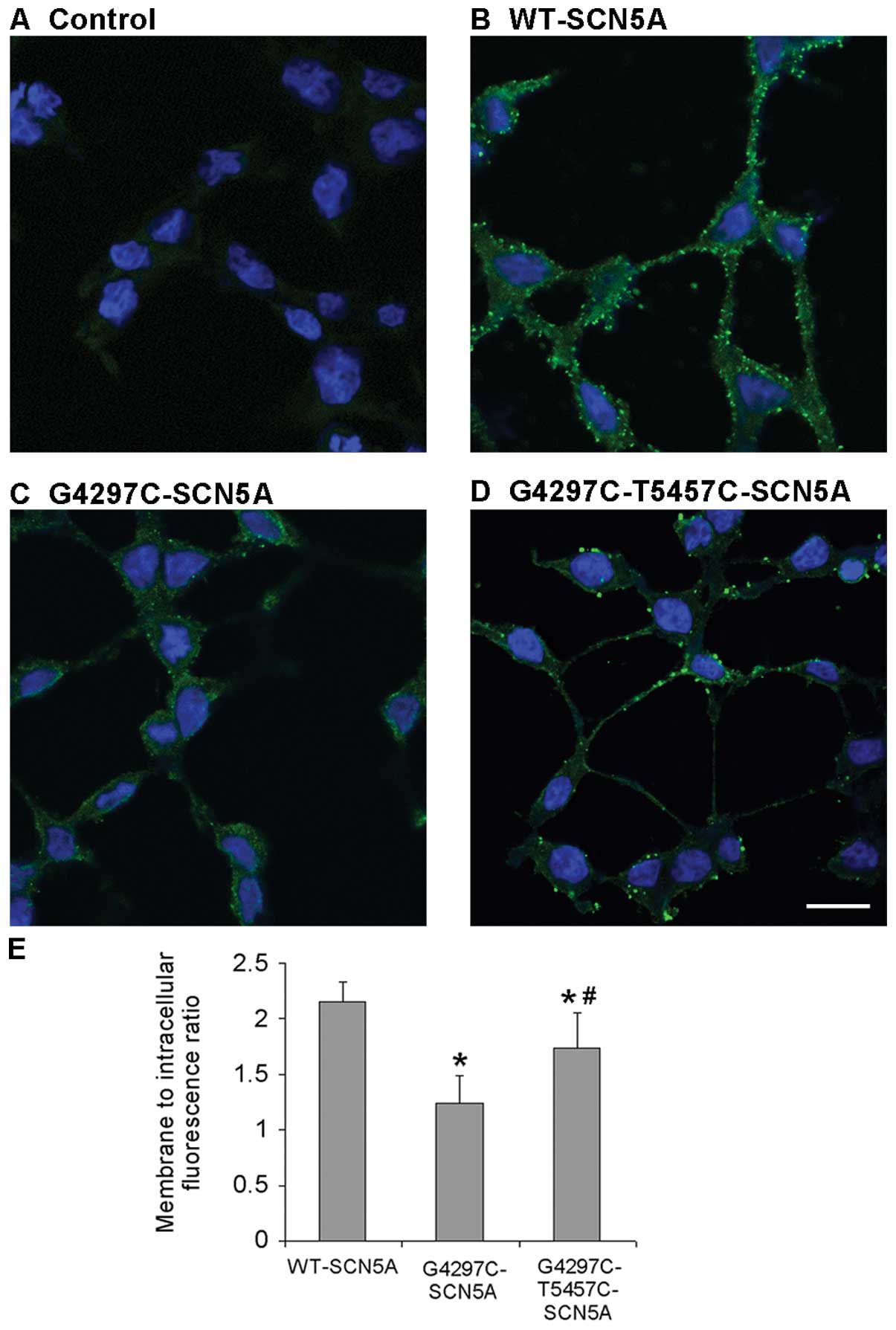
Confocal imaging
To investigate the cellular localization of the WT
and mutant channels, immunocytochemical experiments were performed.
The non-transfected cells displayed background fluorescence
(Fig. 6A). As expected, the
periphery localization was exhibited in cells with WT channels
(Fig. 6B). By contrast, cells
transfected with G4297C mutant channels displayed a very low
expression both in the cell membrane and in the cytoplasm (Fig. 6C). However, the G4297C-T5457C
channels demonstrated a significantly higher expression with a
peripheral localization pattern compared to that of the G4297C
channels, consistent with the increased mRNA levels of
G4297C-T5457C-SCN5A compared with the G4297C channels, which
indicated that the T5457C polymorphism partially rescued the
expression of G4297C mutant channels (Fig. 6D). Summary data depicted in
Fig. 6E revealed that the average
fluorescence intensity per unit area in the membrane divided by the
average fluorescence intensity per unit area in intracellular
compartments (membrane to intracellular fluorescence ratio) was
markedly reduced in the G4297C group compared to that in the WT
group (P=0.0007); however, the ratio significantly increased in the
G4297C-T5457C group compared with the G4297C group (P=0.017) (n=10
for all groups).
Discussion
In this study, we identified an unreported
SCN5A mutation G4297C in a young female patient with
idiopathic VF followed by J waves and ST-segment elevation in the
inferior, lateral and precordial leads demonstrated by documented
ECG. According to the clinical manifestation of the lack of typical
Brugada waves in the right precordial V1 lead, the
patient was not diagnosed as BrS (13,14). Recently, a multicenter study
revealed that J wave and ST-segment elevation in the inferior or
lateral leads, also termed as early repolarization (15,16), accounted for 31% of 206 patients
with idiopathic VF (4). It has
also been reported that up to 11% of patients with BrS presented J
wave and ST-segment elevation in the inferior-lateral leads
associated with an increased number of symptoms (5). Therefore, the nomenclature of ERS or
BrS remains unclear. Based on the new conceptual framework proposed
by Antzelevitch and Yan (1), our
patient may be diagnosed as ERS type 3.
In the present study, we reported a missense G4297C
mutation in the SCN5A gene. After excluding variants in
other genes associated with ERS (6–8)
and BrS (17), including
KCND3, CACNA1C, CACNB2, KCNE3,
SCN1B and KCNJ8, we speculated that the G4297C
mutation in the SCN5A gene was the causative mutation. The
patch-clamp study demonstrated that the G4297C mutation caused a
significant decrease in INa density as well as
altered biophysical characteristics of sodium channels such as
prolonged recovery time from inactivation and depolarizing shift in
activation. The altered properties predisposed the sodium channels
to a ‘loss-of-function’. Further study demonstrated that the mRNA
levels of WT and G4297C mutant channels were similar. However,
immunocytochemistry demonstrated that the G4297C mutant channels
displayed a very low expression both in the cell membrane and in
the cytoplasm. The result suggests that G4297C does not cause a
trafficking defect (18), in
contrast, it affects the translation process or degradation of the
mutant protein. This mechanism differs from three well-known
primary mechanisms that cause a ‘loss-of-function’ of sodium
channels such as i) formation of nonfunctional channels; ii)
altered gating properties; and iii) defective trafficking (18,19). Thus, our findings may represent a
novel mechanism for the ‘loss-of-function’ of sodium channels.
Moreover, several studies found that the R282H
mutation in the SCN5A gene, which leads to BrS, may be fully
rescued by the H558R polymorphism that restores the trafficking of
the mutant protein (20,21). The rescue effect of the synonymous
polymorphism T5457C identified in our study has not yet been
investigated. Our study demonstrated that T5457C did partially
restore the G4297C mutant channels based on the fact that the mRNA
level of the G4297C-T5457C-SCN5A mutant significantly increased
compared to that of the G4297C mutant, corresponding to a
concomitant proportional increase in the number of sodium channels
on the cell surface. A similar phenomenon has been observed in the
ABCC2 gene that encodes the multidrug resistance-associated
protein 2 (MRP2). The synonymous C1146G SNP in the ABCC2
gene increased MRP2 mRNA expression by 2-fold in human livers
(22). However, the essential
mechanism by which a synonymous SNP increases mRNA expression
remains unclear. Several studies have focused on the possibility
that the synonymous SNP alters the stability of mRNA, a phenomenon
suggested by the synonymous C3435T SNP in the ABCB1 gene,
encoding multidrug resistance polypeptide 1 and the synonymous
C971T SNP in the CDSN gene encoding corneodesmosin (23,24). Another explanation was that the
SNP may be in linkage disequilibrium with other polymorphism(s)
acting at the transcriptional level (22).
At present, the transmural voltage gradient mediated
by the transient outward current (Ito) during the
early repolarization phase has been accepted as an explanation of
the pathophysiological mechanism of J wave or ST-segment elevation
on the ECG when the corresponding inward INa was
reduced (25).
Ito was often more prominent in the epicardium of
the right ventricle compared to that of the left ventricle
(26), and therefore, ST-segment
elevation was predisposed to localizing in the right precordial
leads, as noted in BrS. By contrast, in our study, the J waves and
ST-segment elevation of the proband were confined to the inferior,
lateral and left precordial leads. The possible explanation may be
associated with the gender-related difference in
Ito distribution. In the human ventricle, the
transmural Ito gradient is formed by the
differential expression of KChIP2, the gene encoding the β
subunit of the Ito (27). The expression of KChIP2
gene is negatively regulated via the homeodomain Iroquois
transcription factor IRX5 and IRX3 (28). Recently, Gaborit et al
(28) provided evidence that the
expression of KChIP2 is increased in the female left
ventricle, consistent with the decreased expression of IRX3
and IRX5 compared with the right ventricle. The distinction
of expression levels of KChIP2, IRX3 and IRX5
between male and female hearts may cause the J waves and ST-segment
elevation in the inferior, lateral and left precordial leads in
female patients as noted in our proband.
We reported a missense mutation of G4297C in the
SCN5A gene, which caused the ‘loss-of-function’ of sodium
channels, leading to the clinical phenotype of ERS type 3. Our
findings may represent a novel mechanism for the ‘loss-of-function’
of sodium channels by decreasing the number of sodium channels due
to abnormal translation processes. The synonymous T5457C
polymorphism may partially restore the function of the G4297C
mutant channels through the upregulation of mRNA levels. However,
several limitations existed in this study. First, the reasons for
incomplete penetrance in patient I–2 depicted in Fig. 1 were not elucidated. Second,
although the probability was very low, it was unclear whether the
reported mutation and the SNP were on the same allele. The reason
why the T5457C SNP prolonged the recovery from the inactivation of
the sodium channels requires further investigation.
Acknowledgements
The National Basic Research Program of China (973
program, 2007CB512000 and 2007CB512008) provided support to J. Pu
for this research.
References
|
1
|
Antzelevitch C and Yan GX: J wave
syndromes. Heart Rhythm. 7:549–558. 2010. View Article : Google Scholar
|
|
2
|
Miyazaki S, Shah AJ and Haïssaguerre M:
Early repolarization syndrome - a new electrical disorder
associated with sudden cardiac death. Circ J. 74:2039–2044.
2010.PubMed/NCBI
|
|
3
|
Yan GX, Lankipalli RS, Burke JF, Musco S
and Kowey PR: Ventricular repolarization components on the
electrocardiogram: cellular basis and clinical significance. J Am
Coll Cardiol. 42:401–409. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Haïssaguerre M, Derval N, Sacher F, Jesel
L, Deisenhofer I, de Roy L, Pasquié JL, Nogami A, Babuty D,
Yli-Mayry S, et al: Sudden cardiac arrest associated with early
repolarization. N Engl J Med. 358:2016–2023. 2008.PubMed/NCBI
|
|
5
|
Sarkozy A, Chierchia GB, Paparella G,
Boussy T, De Asmundis C, Roos M, Henkens S, Kaufman L, Buyl R,
Brugada R, et al: Inferior and lateral electrocardiographic
repolarization abnormalities in Brugada syndrome. Circ Arrhythm
Electrophysiol. 2:154–161. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Haïssaguerre M, Chatel S, Sacher F,
Weerasooriya R, Probst V, Loussouarn G, Horlitz M, Liersch R,
Schulze-Bahr E, Wilde A, et al: Ventricular fibrillation with
prominent early repolarization associated with a rare variant of
KCNJ8/KATP channel. J Cardiovasc Electrophysiol. 20:93–98.
2009.PubMed/NCBI
|
|
7
|
Medeiros-Domingo A, Tan BH, Crotti L,
Tester DJ, Eckhardt L, Cuoretti A, Kroboth SL, Song C, Zhou Q, Kopp
D, et al: Gain-of-function mutation, S422L, in the KCNJ8-encoded
cardiac K(ATP) channel Kir6.1 as a pathogenic substrate for J-wave
syndromes. Heart Rhythm. 7:1466–1471. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Burashnikov E, Pfeiffer R,
Barajas-Martinez H, Delpón E, Hu D, Desai M, Borggrefe M,
Häissaguerre M, Kanter R, Pollevick GD, et al: Mutations in the
cardiac L-type calcium channel associated with inherited J-wave
syndromes and sudden cardiac death. Heart Rhythm. 7:1872–1882.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Teng S, Gao L, Paajanen V, Pu J and Fan Z:
Readthrough of nonsense mutation W822X in the SCN5A gene can
effectively restore expression of cardiac Na+ channels.
Cardiovasc Res. 83:473–480. 2009. View Article : Google Scholar
|
|
10
|
Hamill OP, Marty A, Neher E, Sakmann B and
Sigworth FJ: Improved patch-clamp techniques for high-resolution
current recording from cells and cell-free membrane patches.
Pflugers Arch. 391:85–100. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Okada T, Ding G, Sonoda H, Kajimoto T,
Haga Y, Khosrowbeygi A, Gao S, Miwa N, Jahangeer S and Nakamura S:
Involvement of N-terminal-extended form of sphingosine kinase 2 in
serum-dependent regulation of cell proliferation and apoptosis. J
Biol Chem. 280:36318–36325. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yao Y, Teng S, Li N, Zhang Y, Boyden PA
and Pu J: Aminoglycoside antibiotics restore functional expression
of truncated HERG channels produced by nonsense mutations. Heart
Rhythm. 6:553–560. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wilde AA, Antzelevitch C, Borggrefe M,
Brugada J, Brugada R, Brugada P, Corrado D, Hauer RN, Kass RS,
Nademanee K, et al: Study Group on the Molecular Basis of
Arrhythmias of the European Society of Cardiology: Proposed
diagnostic criteria for the Brugada syndrome: consensus report.
Circulation. 106:2514–2519. 2002. View Article : Google Scholar
|
|
14
|
Antzelevitch C, Brugada P, Borggrefe M,
Brugada J, Brugada R, Corrado D, Gussak I, LeMarec H, Nademanee K,
Perez Riera AR, et al: Brugada syndrome: report of the second
consensus conference: endorsed by the Heart Rhythm Society and the
European Heart Rhythm Association. Circulation. 111:659–670. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shu J, Zhu T, Yang L, Cui C and Yan GX:
ST-segment elevation in the early repolarization syndrome,
idiopathic ventricular fibrillation, and the Brugada syndrome:
cellular and clinical linkage. J Electrocardiol. 38(Suppl 4):
S26–S32. 2005. View Article : Google Scholar
|
|
16
|
Di Grande A, Tabita V, Lizzio MM,
Giuffrida C, Bellanuova I, Lisi M, Le Moli C and Amico S: Early
repolarization syndrome and Brugada syndrome: is there any linkage?
Eur J Intern Med. 19:236–240. 2008.PubMed/NCBI
|
|
17
|
Hedley PL, Jørgensen P, Schlamowitz S,
Moolman-Smook J, Kanters JK, Corfield VA and Christiansen M: The
genetic basis of Brugada syndrome: a mutation update. Hum Mutat.
30:1256–1266. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Baroudi G, Pouliot V, Denjoy I, Guicheney
P, Shrier A and Chahine M: Novel mechanism for Brugada syndrome:
defective surface localization of an SCN5A mutant (R1432G). Circ
Res. 88:E78–E83. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Baroudi G, Acharfi S, Larouche C and
Chahine M: Expression and intracellular localization of an SCN5A
double mutant R1232W/T1620M implicated in Brugada syndrome. Circ
Res. 90:E11–E16. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Poelzing S, Forleo C, Samodell M, Dudash
L, Sorrentino S, Anaclerio M, Troccoli R, Iacoviello M, Romito R,
Guida P, et al: SCN5A polymorphism restores trafficking of a
Brugada syndrome mutation on a separate gene. Circulation.
114:368–376. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gui J, Wang T, Trump D, Zimmer T and Lei
M: Mutation-specific effects of polymorphism H558R in SCN5A-related
sick sinus syndrome. J Cardiovasc Electrophysiol. 21:564–573. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Niemi M, Arnold KA, Backman JT, Pasanen
MK, Gödtel-Armbrust U, Wojnowski L, Zanger UM, Neuvonen PJ,
Eichelbaum M, Kivistö KT and Lang T: Association of genetic
polymorphism in ABCC2 with hepatic multidrug resistance-associated
protein 2 expression and pravastatin pharmacokinetics.
Pharmacogenet Genomics. 16:801–808. 2006. View Article : Google Scholar
|
|
23
|
Wang D, Johnson AD, Papp AC, Kroetz DL and
Sadée W: Multidrug resistance polypeptide 1 (MDR1, ABCB1) variant
3435C>T affects mRNA stability. Pharmacogenet Genomics.
15:693–704. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Capon F, Allen MH, Ameen M, Burden AD,
Tillman D, Barker JN and Trembath RC: A synonymous SNP of the
corneodesmosin gene leads to increased mRNA stability and
demonstrates association with psoriasis across diverse ethnic
groups. Hum Mol Genet. 13:2361–2368. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yan GX and Antzelevitch C: Cellular basis
for the Brugada syndrome and other mechanisms of arrhythmogenesis
associated with ST-segment elevation. Circulation. 100:1660–1666.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Di Diego JM, Cordeiro JM, Goodrow RJ, Fish
JM, Zygmunt AC, Pérez GJ, Scornik FS and Antzelevitch C: Ionic and
cellular basis for the predominance of the Brugada syndrome
phenotype in males. Circulation. 106:2004–2011. 2002.PubMed/NCBI
|
|
27
|
Rosati B, Pan Z, Lypen S, Wang HS, Cohen
I, Dixon JE and McKinnon D: Regulation of KChIP2 potassium channel
beta subunit gene expression underlies the gradient of transient
outward current in canine and human ventricle. J Physiol.
533:119–125. 2001. View Article : Google Scholar
|
|
28
|
Gaborit N, Varro A, Le Bouter S, Szuts V,
Escande D, Nattel S and Demolombe S: Gender-related differences in
ion-channel and transporter subunit expression in non-diseased
human hearts. J Mol Cell Cardiol. 49:639–646. 2010. View Article : Google Scholar : PubMed/NCBI
|