Introduction
Pulmonary vascular remodeling is one of the most
important pathological changes in patients with pulmonary arterial
hypertension (PAH). Endothelial cells are recognized as major
regulators of vascular function. The balance between endothelial
cell survival and death is critical in various processes, such as
the regulation of vasoconstriction and vasodilation, smooth muscle
cell growth and migration, thrombotic formation, intravascular
inflammation and vascular remodeling (1,2).
Inflammation is one of the main features of PAH, and circulating
levels of cytokines, including tumor necrosis factor-α (TNF-α), are
elevated in patients with PAH (3,4).
During the early stages of vascular remodeling, inflammatory
infiltration directly participates in the apoptosis of endothelial
cells. Considerable experimental evidence suggests that the
apoptosis of endothelial cells induces the release of mediators, in
particular, transforming growth factor-β1 (TGF-β1), which induces
the proliferation and migration of vascular smooth muscle cells
(5,6). Sturrock et al (7) demonstrated that TGF-β1 is abundantly
expressed in patients with pulmonary hypertension, and promotes
pulmonary artery smooth muscle cell (PASMC) proliferation in low
serum medium. The inhibition of TGF-β1 signaling has been
demonstrated to attenuate pulmonary vascular remodeling and
increase right ventricular pressure in animal models (8).
Arachidonic acid (AA) is an essential
polyunsaturated fatty acid which is esterified to membrane
phospholipids. Four epoxyeicosatrienoic acid (EET) regioisomers
(5,6-, 8,9-, 11,12- and 14,15-EET) are the metabolites of AA by the
cytochrome 450 (CYP) monooxygenase pathway (9,10).
Similar to many eicosanoids, EETs have multiple biological
functions, including the regulation of cardiovascular inflammation,
the reduction of blood pressure, as well as anti-atherosclerotic
functions in multiple model systems. EETs inhibit endothelial
activation and leukocyte adhesion by suppressing nuclear factor
(NF)-κB activation (11), thus
activating peroxisome proliferator-activated receptor (PPAR)-α and
PPAR-γ (12–14), and increasing heme oxygenase
(HO)-1 expression (15). In
addition to their potent vasodilatory action and anti-inflammatory
effects, EETs have been associated with a number of physiological
functions, such as vascular protection and cardiovascular
homeostasis. EETs upregulate endothelial nitric oxide synthase
(eNOS) expression and activity (16), and stimulate angiogenesis and
endothelial cell growth (17,18).
Evidence suggests that EETs exert many protective
effects on systemic vascular homeostasis (19). However, the precise mechanisms
responsible for the effects of EETs on pulmonary vascular
homeostasis remain to be elucidated. Our group recently
demonstrated the protective effects of gene delivery with
cytochrome P450 2J2 (CYP2J2) on monocrotaline-induced PAH in rats
(20). In the present study,
porcine pulmonary artery endothelial cells (PAECs) and PASMCs were
used to evaluate the effects of exogenous EETs or CYP2J2
overexpression on TNF-α-induced PAEC apoptosis and TGF-β1-induced
PASMC proliferation and migration. In other words, this model was
utilized to examine the hypothesis that exogenous EETs or CYP2J2
overexpression may attenuate pulmonary vascular remodeling.
Materials and methods
Experimental reagents
FBS, DMEM and trypsin were purchased from Invitrogen
(Carlsbad, CA, USA). Collagenase II and the cell counting kit-8
(CCK-8) were purchased from Beyotime Biotechnology (Shanghai,
China). TNF-α, TGF-β1, phosphoinositide 3-kinase (PI3K) inhibitor
(LY294002), mitogen-activated protein kinase (MAPK) inhibitor
(apigenin) and extracellular signal-regulated kinase (ERK)
inhibitor (PD98059) were obtained from Sigma-Aldrich (St. Louis,
MO, USA). EETs were from Cayman Chemical Co. (Ann Arbor, MI, USA).
Antibody against CYP2J2 was purchased from Abcam Inc. (Cambridge,
MA, USA). Antibody directed against PI3K was obtained from Cell
Signaling Technology, Inc. (Beverly, MA, USA). Other antibodies
used in this study were from Santa Cruz Biotechnology, Inc. (Santa
Cruz, CA, USA). The Annexin V-FITC Apoptosis Detection Kit was
purchased from KeyGen Biotech, Co. (Nanjing, China), and the
Annexin V PE Apoptosis Detection Kit was from eBioscience, Inc.
(San Diego, CA, USA). All other reagents were purchased from
standard commercial suppliers unless otherwise indicated.
Cell culture and experimental
incubation
PAECs and PASMCs were cultured from porcine
pulmonary arteries as previously described (18,21). PAECs and PASMCs, which were used
between passages 2 and 6, were isolated from porcine pulmonary
arteries obtained from a local abattoir within 1 h of slaughter.
Endothelial cells were digested with 1.7 mg/ml collagenase II, then
the pulmonary arteries were cut open along the longitudinal axis,
and the residual endothelium was gently removed by scraping the
luminal surface and washed away with phosphate-buffered saline
(PBS). The adventitia layer was removed by blunt dissection, and
the medial smooth muscle tissue was minced into 1 mm3
explant pieces. PAECs and the explant pieces of smooth muscle were
maintained in Dulbecco's modified Eagle's medium (DMEM)
supplemented with 10% fetal bovine serum (FBS), penicillin (100
U/ml), streptomycin (100 U/ml), 5 mM L-glutamine and 1.5 g/l sodium
bicarbonate. The medium was changed every 2 days. The cells were
cultured in an incubator at 37ºC and maintained in a humidified
atmosphere with 5% CO2. The purity and identity of the
endothelial cells and smooth muscle cells were verified by their
typical morphological patterns and by immunofluorescent staining
for factor VIII-related antigen and α-smooth muscle actin (α-SMA),
respectively (16,21). All passages were performed using
0.05% trypsin and 0.02% EDTA. Cell viability was assessed using the
CCK-8 assay.
Treatment of PAECs and PASMCs
Prior to each experiment, the PAECs and PASMCs were
quiesced for 24 h in DMEM without serum. The PASMCs were treated
with TGF-β1 in low serum (0.4%), and PAECs were treated with TNF-α
in 10% FBS in the presence/absence of a physiologically relevant
concentration [as previously described (22)] of EETs (250 nM) for 24 h.
Alternatively, the PAECs were infected with recombinant
adeno-associated viral vector (rAAV)-green fluorescent protein
(GFP) or rAAV-CYP2J2 for 5 days to obtain maximal expression and
then incubated with TNF-α. To investigate the anti-apoptotic
effects of CYP2J2 or EETs and the possible signaling mechanisms
involved, the infected PAECs were treated for 45 min with/without
PI3K inhibitor (LY294002, 20 μM), ERK inhibitor (PD98059, 20 μM) or
MAPK inhibitor (apigenin, 20 μM), followed by incubation with TNF-α
(10 ng/ml) for 24 h. Alternatively, following pre-treatment
with/without LY294002, PD98059 or apigenin, the cells were
incubated with 14,15-EET (250 nM) for 30 min prior to treatment
with TNF-α.
Recombinant adeno-associated virus
The PAECs were infected with rAAV-CYP2J2 or rAAV-GFP
(~50 virions/cell), packed and purified as previously described
(16,18), and grown for 5 days to obtain
maximal expression. Recombinant adeno-associated viral vectors
containing GFP or CYP2J2, pcDNA3.1-GFP plasmids and pcDNA3.1-CYP2J2
plasmids were prepared as previously described (23). The transfection efficiency of
rAAV-GFP was detected by flow cytometry and fluorescence microscopy
at 5 days post-infection.
Cell viability assay
Cell viability was measured using the CCK-8 assay,
in which cellular dehydrogenase activity in the living cells was
detected. Cell viability was expressed as a percentage of the value
in the untreated control culture (24,25). All experiments were performed in
triplicate on 3 separate occasions.
Flow cytometric analysis of
apoptosis
As previously described (26,27), apoptotic responses were assessed
by flow cytometry (Annexin V-FITC, propidium iodide and binding
buffer) following the treatment of PAECs with 14,15-EET. As the
emission wavelength of GFP and Annexin V-FITC are similar, we used
the eBioscience Annexin V PE apoptosis detection kit (Annexin V-PE,
7-AAD viability staining solution, and binding buffer) to detect
the apoptosis of virus-infected PAECs. All flow cytometry analyses
were performed using commercially-available CellQuest software (BD
Biosciences, San Jose, CA, USA).
Western blot analysis
The total expression levels and phosphorylation
levels of signal transduction molecules were measured by western
blot analysis as previously described (20). Protein expression was quantified
by densitometry and normalized to β-actin expression.
Ki-67 staining
The PASMCs were seeded on a 12-well plate to 80%
confluence, then incubated with TGF-β1 and EETs as indicated above.
After 24 h, the PASMCs were probed with anti-Ki-67 antibody as
described previously (28).
Hematoxylin was used to counterstain the nuclei. In all assays,
negative controls were prepared using PBS instead of anti-Ki-67
antibody in order to exclude non-specific staining.
Cell migration assay
Migration assay was performed using Transwell
chambers (Corning, Lowell, MA, USA) with a pore size of 8 μm
(19,29,30). Briefly, the PASMCs were suspended
in DMEM with 0.4% FBS at a concentration of 5×105
cells/ml, and 0.2 ml aliquots of the cell suspension
(1×105 cells) were added to the upper chambers. The
cells in the upper chamber were incubated with EETs at 37ºC for 30
min, then TGF-β1 was added to the medium of the lower chamber that
contained 0.6 ml of DMEM, 0.4% FBS. The migration lasted for 4 h at
37ºC in a CO2 incubator. In order to determine the
number of migrated cells, the cells on the upper surface of the
filters were carefully scraped off with a cotton swab. The cells
that had migrated through the filters were fixed to the membrane
using 4% paraformaldehyde for 30 min, then stained with 0.1%
crystal violet for 10 min at room temperature, and finally examined
and photographed under a microscope (x200 magnification). The
quantification of migrated cells was performed (5 randomly selected
fields/Transwell from at least 3 Transwells/experiment).
Statistical analysis
All data was presented as the means ± SEM.
Statistically significant differences between 2 groups were
calculated using the Student's t-test, and one-way ANOVA was used
to certify statistical differences among 3 groups; a P-value
<0.05 was considered to indicate a statistically significant
difference.
Results
Effects of exogenous treatment with EETs
on viability of PAECs and PASMCs
Proliferation and apoptosis were measured using the
CCK-8 assay. We wished to determine the effects of exogenous EETs
on the viability of PAECs and PASMCs by comparing the responses of
the 2 primary cell types to apoptosis or proliferation. In the
PAECs, the physiologically relevant concentration of EETs (250 nM)
had no significant effects on the cell viability of the control
PAECs (Fig. 1A). TNF-α (5–10
ng/ml) induced a significant reduction in the number of viable
cells (Fig. 1B), and EETs
reversed the reduction in TNF-α (10 ng/ml)-induced cell viability
(Fig. 1C). In the PASMCs, TGF-β1
(5–10 ng/ml) induced a significant increase in the number of viable
cells (Fig. 1D). However, EETs
(250 nM) did not reverse the increase in TGF-β1-induced cell
viability (Fig. 1E). These data
suggest that EETs inhibit PAEC apoptosis, but do not suppress PASMC
proliferation.
EETs and CYP2J2 overexpression inhibits
TNF-α-induced apoptosis of PAECs
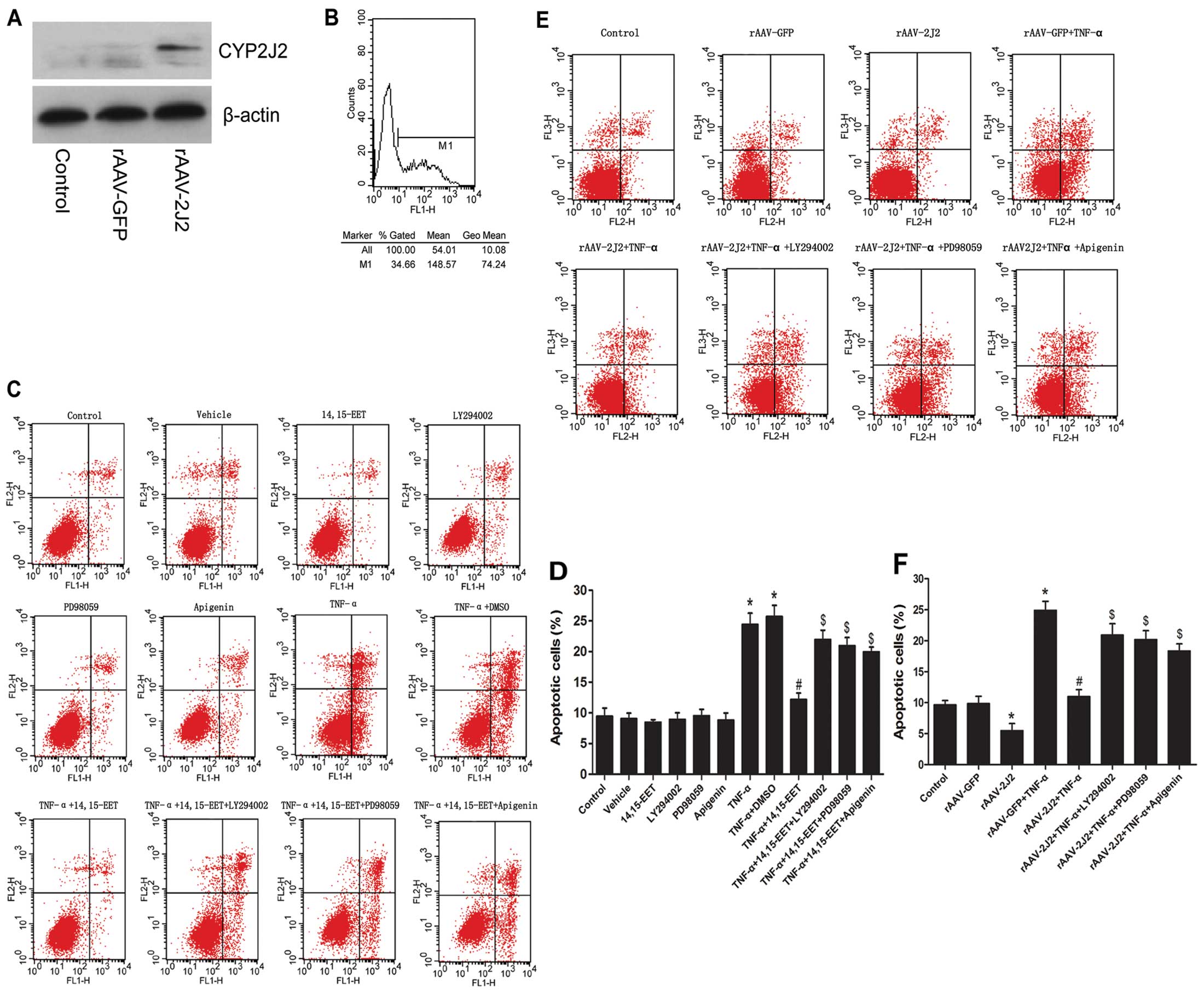
To determine the transfection efficiency of
rAAV-CYP2J2 in PAECs, the uptake of fluorescently labeled rAAV-GFP
was detected by flow cytometry at 5 days post-transfection. The
results revealed a high transfection efficiency (>34% cells
displayed green fluorescence with rAAV-GFP) (Fig. 2B). The protein expression of
CYP2J2 was examined by western blot analysis (Fig. 2A). CYP2J2 expression induced a
marked increase in the protein levels in the rAAV-CYP2J2 group
compared with rAAV-GFP. We also evaluated the effects of exogenous
EETs and CYP2J2 overexpression on the apoptosis of PAECs by flow
cytometry. Our results indicated that 14,15-EET partially abolished
the apoptosis of PAECs induced by TNF-α (Fig. 2C and D). Likewise, the rAAV-CYP2J2
+ TNF-α group showed significantly reduced cell apoptosis compared
with the rAAV-GFP + TNF-α group (Fig.
2E and F). The anti-apoptotic effects of 14,15-EET or CYP2J2
overexpression were attenuated by LY294002 (PI3K inhibitor),
PD98059 (ERK inhibitor) or apigenin (MAPK inhibitor) (Fig. 2C–F). These results demonstrate
that the anti-apoptotic effects of EETs and CYP2J2 overexpression
are mediated, at least in part, through the activation of PI3K, ERK
and MAPK.
Effects of 14,15-EET and CYP2J2
overexpression on apoptosis-regulating protein expression in
PAECs
The incubation of PAECs with a physiologically
relevant concentration of 14,15-EET had no effects on the baseline
levels of Bcl-2, Bcl-xL and Bax, but significantly attenuated the
TNF-α-induced downregulation of Bcl-2 and Bcl-xL expression, and
prevented the upregulation of Bax (Fig. 3A and B). The overexpression of
CYP2J2 in the PAECs induced a significant increase in the
expression Bcl-2 and Bcl-xL, as well as a significant decrease in
the baseline levels of Bax. Moreover, it also significantly
inhibited the TNF-α-induced downregulation of Bcl-2, Bcl-xL and the
TNF-α-induced upregulation of Bax (Fig. 3C and D). Furthermore, the effects
of 14,15-EET and CYP2J2 overexpression were attenuated by LY294002,
PD98059 or apigenin (Fig. 3),
supporting the role of the ERK and PI3K/Akt signaling pathways in
the EET-mediated regulation of apoptosis.
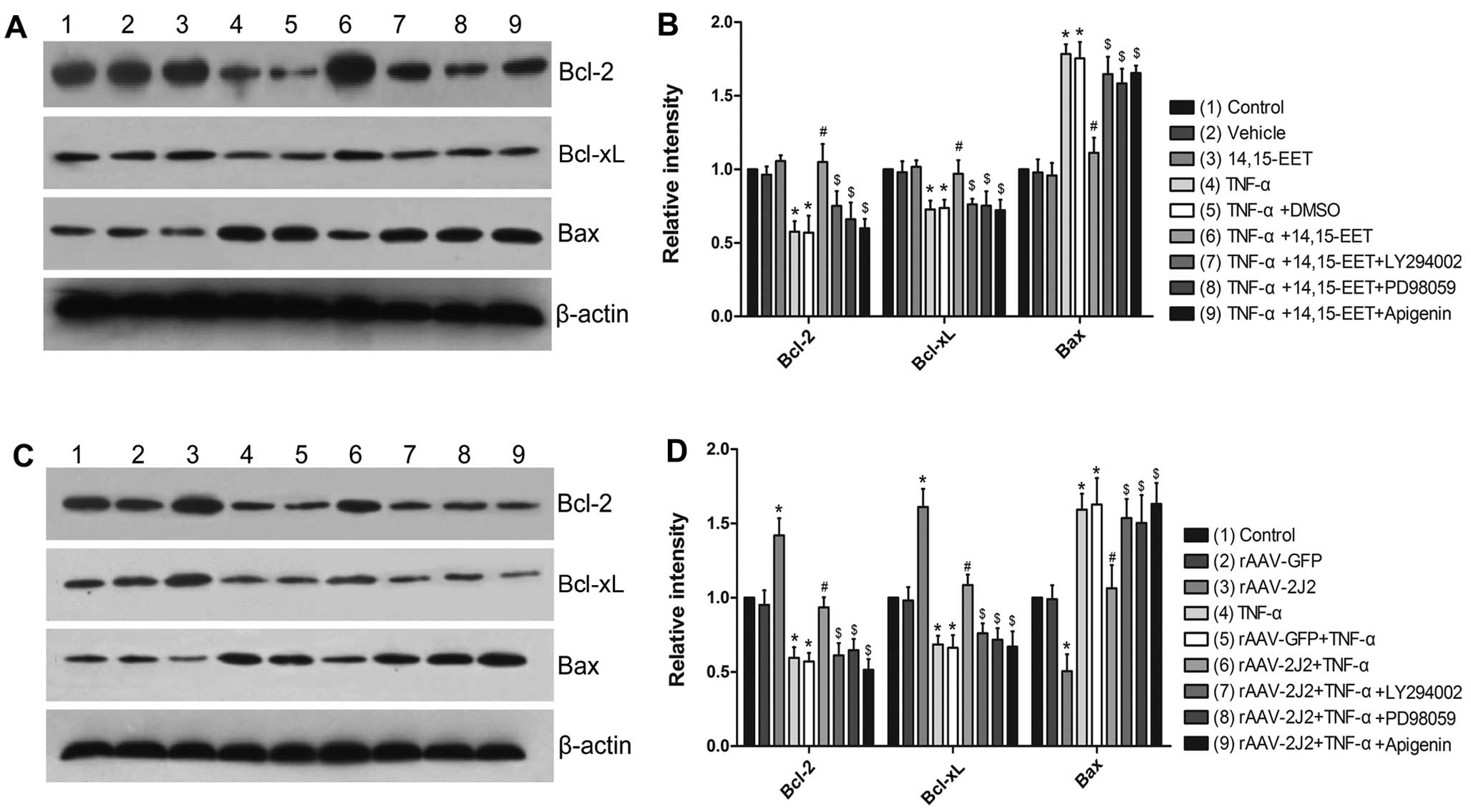 | Figure 3Effects of 14:15-EET and CYP2J2
overexpression on apoptosis-regulating protein expression in
pulmonary artery endothelial cells (PAECs). (A and B)
Representative western blots and densitometry results showing
altered levels of Bcl-2, Bcl-xL and Bax following the exogenous
administration of 14,15-EET, and the effects of LY294002, PD98059
or apigenin. (C and D) Representative western blots and
densitometry results showing the altered levels of Bcl-2, Bcl-xL
and Bax following CYP2J2 overexpression, and the effects of
LY294002, PD98059 or apigenin (*P<0.05 vs. control or
vs. rAAV-GFP; #P<0.05 vs. TNF-α + DMSO or vs.
rAAV-GFP + TNF-α; $P<0.05 vs. TNF-α + 14:15-EET or
vs. rAAV-CYP2J2 + TNF-α). TNF-α, tumor necrosis factor-α; rAAV,
recombinant adeno-associated viral vector; GFP, green fluorescent
protein. |
Effects of 14,15-EET and CYP2J2
overexpression on PI3K-dependent Akt phosphorylation and ERK1/2
pathway in PAECs
The treatment of PAECs with TNF-α inhibited the
expression of PI3K and PI3K-dependent Akt phosphorylation;
14,15-EET did not significantly increase PI3K protein expression
and Akt phosphorylation in the control cells, whereas 14,15-EET
significantly increased PI3K protein expression and Akt
phosphorylation in the presence of TNF-α (Fig. 4A and B). The effects of 14,15-EET
were inhibited when the cells were pre-treated with LY294002 (a
PI3K inhibitor) (Fig. 4A and B).
Similarly, the overexpression of CYP2J2 significantly increased the
expression of PI3K protein and Akt phosphorylation in the presence
and absence of TNF-α, compared with the uninfected and
rAAV-GFP-infected control cells. This effect was blocked by
LY294002 (Fig. 4C and D). TNF-α
also significantly reduced the phosphorylation of ERK1/2 in the
PAECs. 14,15-EET did not significantly induce ERK1/2
phosphorylation, while 14,15-EET significantly induced ERK1/2
phosphorylation in the presence of TNF-α (Fig. 4A and B). The overexpression of
CYP2J2 significantly increased ERK1/2 phosphorylation in the
presence and absence of TNF-α (Fig.
4C and D). Furthermore, the effects of 14,15-EET and CYP2J2
overexpression on ERK1/2 phosphorylation were inhibited by PD98059
(an ERK inhibitor) and apigenin (a MAPK inhibitor) (Fig. 4C and D). Taken together, these
results suggest that exogenous 14,15-EET and CYP2J2 overexpression
activates the PI3K/Akt and ERK signaling pathways, which may
contribute to the anti-apoptotic effects observed in the PAECs.
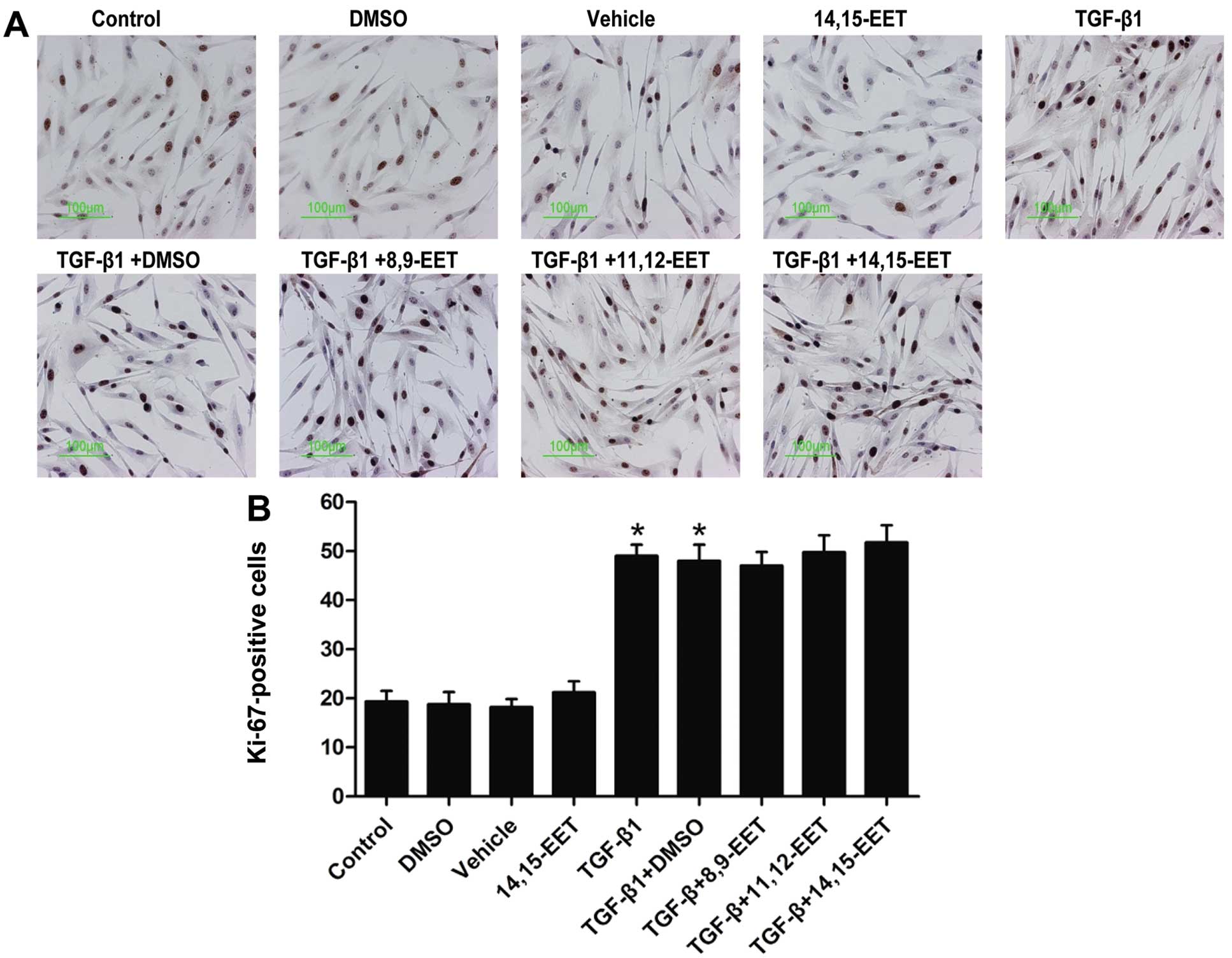
Effect of Ki-67 staining following
stimulation of PASMCs with TGF-β1
There was a small percentage (~20%) of PASMCs
entering the cell cycle in the untreated control cell culture
(Fig. 5). However, there was a
significant increase (~50%) in the number of Ki-67-positive cells
after TGF-β1 (10 ng/ml) was administered for 24 h. However, EETs
(250 nM) did not reverse the increase in the number of
TGF-β1-induced Ki-67-positive cells (Fig. 5). These findings suggest that EETs
does not suppress PASMC proliferation.
Effect of EETs on TGF-β1-induced PASMC
migration
To investigate the effects of EETs on TGF-β1-induced
PASMC migration, a modified Transwell apparatus was used. The
stimulation of the PASMCs with TGF-β1 (10 ng/ml) caused a ~3-fold
increase in PASMC migration (Fig.
6B). At a concentration of 250 nM, 11,12-EET and 14,15-EET
significantly inhibited the TGF-β1-induced PASMC migration. By
contrast, 8,9-EET had no significant effect (Fig. 6B). These findings suggest that
11,12-EET and 14,15-EET attenuate the migration potential of PASMCs
in vitro.
Discussion
Pulmonary vascular remodeling is characterized by
endothelial dysfunction, abnormal muscularization of pulmonary
arterioles, the upregulation of inflammatory cytokines and
leukocyte infiltration (31,32). Inflammation plays an important
role in various types of human PAH (33). TNF-α has been demonstrated as a
key pro-inflammatory cytokine in severe pulmonary hypertension.
Fujita et al (34)
demonstrated that the overexpression of TNF-α results in severe
pulmonary hypertension in mice. During the early stages of vascular
remodeling, inflammatory cells infiltration directly participates
in the apoptosis of endothelial cells. A body of evidence has
demonstrated that the apoptosis of endothelial cells is an
initiating mechanism for pulmonary vascular remodeling, and
directly leads to microvascular obliteration (35,36). Moreover, increasing evidence
suggests that pulmonary vascular remodeling may be reversed by
inhibiting the apoptosis of endothelial cells during the early
stages of PAH (37–39). In another study, Sakao et
al (6) reported that the
apoptosis of endothelial cells induces the release of mediators, in
particular TGF-β1, which activates the proliferation and migration
of vascular smooth muscle cells. Liu et al (40) showed that TGF-β1 increased the
progression of cells from the G0/G1 phase to the G2/M + S phase and
regulated the cell cycle progression of PASMCs. Considerable
experimental evidence suggests that TGF-β1 mediates human PASMC
proliferation in persistent hypoxia-induced PAH (41). However, it is not clear whether
EETs exert anti-apoptotic effects on PAECs, particularly in the
case of apoptosis induced by inflammatory cytokines (such as
TNF-α). Moreover, the anti-proliferative and anti-migratory effects
of EET on PASMCs remain elusive. Thus, in this study, we sought to
evaluate the potential effects of exogenous EETs and CYP2J2
overexpression on TNF-α-induced PAEC apoptosis and TGF-β1-induced
PASMC proliferation and migration.
Our findings demonstrated that EETs significantly
reversed the TNF-α-induced reduction in cell viability. Moreover,
EETs and CYP2J2 overexpression effectively protected the PAECs from
TNF-α-induced apoptosis, as evaluated by FACS analysis. EETs and
CYP2J2 overexpression significantly inhibited the downregulation of
TNF-α-induced Bcl-2 and Bcl-xL protein expression, and the
upregulation of TNF-α-induced Bax protein expression. These effects
were attenuated by the addition of LY294002 (a PI3K inhibitor),
PD98059 (an ERK inhibitor) and apigenin (a MAPK inhibitor).
Furthermore, EETs and CYP2J2 overexpression significantly induced
PI3K protein expression, Akt phosphorylation and ERK1/2
phosphorylation in the presence of TNF-α, and these effects were
attenuated by the addition of LY294002, PD98059 and apigenin,
respectively. These anti-apoptotic effects were significantly
attenuated by the inhibition of the PI3K/Akt and MAPK signaling
pathways, suggesting that the anti-apoptotic effects of EETS are
mediated, at least in part, through the activation of the PI3K/Akt
and MAPK signaling pathways.
The monocrotaline-induced primary pulmonary
hypertension model, which has previously been used by Revermann
et al (42), is highly
toxic to endothelial cells in the lungs. It induced endothelial
cell apoptosis, vascular inflammation, cellular proliferation and
vascular remodeling. We have previously reported that CYP2J2
overexpression, which is known to increase EET biosynthesis,
significantly ameliorated monocrotaline-induced PAH in rats
(20). We found that CYP2J2
overexpression attenuated right ventricle systolic pressure (RVSP)
and hypertrophy of the right ventricle (RV) and pulmonary vessel
walls in vivo. However, our results did not favor any role
of the anti-proliferative effects of EETs on TGF-β1-induced PASMCs
in vitro, indicating that a more complex mechanism of
inflammation may be present for PASMC proliferation. These results
were supported by those from previous studies in cultured rat
PASMCs, showing that 11,12-EET and soluble epoxide hydrolase (sEH)
inhibition have no effect on platelet-derived growth factor (PDGF),
serum or serotonin-induced smooth muscle cell proliferation
(19). In addition, we also
discovered that at a concentration of 250 nM 11,12-EET and
14,15-EET significantly inhibited the TGF-β1-induced PASMC
migration. Taken together, these data indicate that EETs suppress
pulmonary vascular remodeling mainly through the inhibition of
PASMC migration, and not by inhibiting proliferation; it could also
be speculated that the anti-proliferative effects of EETs in
vivo are indirect in nature.
In conclusion, the present study reveals a novel
role for the protective effects of EETs against pulmonary vascular
remodeling. EETs exerted potent anti-apoptotic effects, markedly
attenuating the TNF-α-induced apoptosis of pulmonary arterial
endothelial cells. Two mechanisms were found to be involved in
these important protective effects: firstly, EETs and CYP2J2
overexpression inhibited the decrease in the expression of the
anti-apoptotic proteins, Bcl-2 and Bcl-xL, and the increase in the
expression of the pro-apoptotic protein, Bax, mediated by TNF-α;
secondly, EETs activated the PI3K/Akt and ERK signaling pathways.
In addition, EETs inhibited the TGF-β1-induced PASMC migration. Our
findings suggest that EETs may be a novel approach to the treatment
of pulmonary vascular remodeling complications in diseases such as
PAH.
Acknowledgments
This study was supported by grants from the National
Natural Science Foundation of China (nos. 30971247 and
81170111).
References
|
1
|
Stefanec T: Endothelial apoptosis: could
it have a role in the pathogenesis and treatment of disease? Chest.
117:841–854. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yang X, Long L, Reynolds PN and Morrell
NW: Expression of mutant BMPR-II in pulmonary endothelial cells
promotes apoptosis and a release of factors that stimulate
proliferation of pulmonary arterial smooth muscle cells. Pulm Circ.
1:103–110. 2011. View Article : Google Scholar
|
|
3
|
Wort SJ, Ito M, Chou PC, et al:
Synergistic induction of endothelin-1 by tumor necrosis factor
alpha and interferon gamma is due to enhanced NF-kappaB binding and
histone acetylation at specific kappaB sites. J Biol Chem.
284:24297–24305. 2009. View Article : Google Scholar
|
|
4
|
Soon E, Holmes AM, Treacy CM, et al:
Elevated levels of inflammatory cytokines predict survival in
idiopathic and familial pulmonary arterial hypertension.
Circulation. 122:920–927. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Antonelli-Orlidge A, Saunders KB, Smith SR
and D'Amore PA: An activated form of transforming growth factor
beta is produced by cocultures of endothelial cells and pericytes.
Proc Natl Acad Sci USA. 86:4544–4548. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sakao S, Taraseviciene-Stewart L, Wood K,
Cool CD and Voelkel NF: Apoptosis of pulmonary microvascular
endothelial cells stimulates vascular smooth muscle cell growth. Am
J Physiol Lung Cell Mol Physiol. 291:L362–L368. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sturrock A, Cahill B, Norman K, et al:
Transforming growth factor-beta1 induces Nox4 NAD(P)H oxidase and
reactive oxygen species-dependent proliferation in human pulmonary
artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol.
290:L661–L673. 2006. View Article : Google Scholar
|
|
8
|
Ma W, Han W, Greer PA, et al: Calpain
mediates pulmonary vascular remodeling in rodent models of
pulmonary hypertension, and its inhibition attenuates pathologic
features of disease. J Clin Invest. 121:4548–4566. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wu S, Moomaw CR, Tomer KB, Falck JR and
Zeldin DC: Molecular cloning and expression of CYP2J2, a human
cytochrome P450 arachidonic acid epoxygenase highly expressed in
heart. J Biol Chem. 271:3460–3468. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zeldin DC: Epoxygenase pathways of
arachidonic acid metabolism. J Biol Chem. 276:36059–36062. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Node K, Huo Y, Ruan X, et al:
Anti-inflammatory properties of cytochrome P450 epoxygenase-derived
eicosanoids. Science. 285:1276–1279. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pasceri V, Wu HD, Willerson JT and Yeh ET:
Modulation of vascular inflammation in vitro and in vivo by
peroxisome proliferator-activated receptor-gamma activators.
Circulation. 101:235–238. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang N, Verna L, Chen NG, et al:
Constitutive activation of peroxisome proliferator-activated
receptor-gamma suppresses pro-inflammatory adhesion molecules in
human vascular endothelial cells. J Biol Chem. 277:34176–34181.
2002. View Article : Google Scholar
|
|
14
|
Feige JN, Gelman L, Michalik L, Desvergne
B and Wahli W: From molecular action to physiological outputs:
peroxisome proliferator-activated receptors are nuclear receptors
at the crossroads of key cellular functions. Prog Lipid Res.
45:120–159. 2006. View Article : Google Scholar
|
|
15
|
Sacerdoti D, Colombrita C, Di Pascoli M,
et al: 11,12-epoxyeicosatrienoic acid stimulates heme-oxygenase-1
in endothelial cells. Prostaglandins Other Lipid Mediat.
82:155–161. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang H, Lin L, Jiang J, et al:
Up-regulation of endothelial nitric-oxide synthase by
endothelium-derived hyperpolarizing factor involves
mitogen-activated protein kinase and protein kinase C signaling
pathways. J Pharmacol Exp Ther. 307:753–764. 2003. View Article : Google Scholar
|
|
17
|
Wang Y, Wei X, Xiao X, et al: Arachidonic
acid epoxygenase metabolites stimulate endothelial cell growth and
angiogenesis via mitogen-activated protein kinase and
phosphatidylinositol 3-kinase/Akt signaling pathways. J Pharmacol
Exp Ther. 314:522–532. 2005. View Article : Google Scholar
|
|
18
|
Yang S, Lin L, Chen JX, et al: Cytochrome
P-450 epoxygenases protect endothelial cells from apoptosis induced
by tumor necrosis factor-alpha via MAPK and PI3K/Akt signaling
pathways. Am J Physiol Heart Circ Physiol. 293:H142–H151. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sun J, Sui X, Bradbury JA, Zeldin DC,
Conte MS and Liao JK: Inhibition of vascular smooth muscle cell
migration by cytochrome p450 epoxygenase-derived eicosanoids. Circ
Res. 90:1020–1027. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zheng C, Wang L, Li R, et al: Gene
delivery of cytochrome p450 epoxygenase ameliorates
monocrotaline-induced pulmonary artery hypertension in rats. Am J
Respir Cell Mol Biol. 43:740–749. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tian X, Vroom C, Ghofrani HA, et al:
Phosphodiesterase 10A upregulation contributes to pulmonary
vascular remodeling. PLoS One. 6:e181362011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dhanasekaran A, Al-Saghir R, Lopez B, et
al: Protective effects of epoxyeicosatrienoic acids on human
endothelial cells from the pulmonary and coronary vasculature. Am J
Physiol Heart Circ Physiol. 291:H517–H531. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yang B, Graham L, Dikalov S, et al:
Overexpression of cytochrome P450 CYP2J2 protects against
hypoxia-reoxygenation injury in cultured bovine aortic endothelial
cells. Mol Pharmacol. 60:310–320. 2001.PubMed/NCBI
|
|
24
|
Yuan P, Salvadore G, Li X, et al:
Valproate activates the Notch3/c-FLIP signaling cascade: a strategy
to attenuate white matter hyperintensities in bipolar disorder in
late life? Bipolar Disord. 11:256–269. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kim HY, Hwang JY, Kim SW, et al: The CXCR4
antagonist AMD3100 has dual effects on survival and proliferation
of myeloma cells in vitro. Cancer Res Treat. 42:225–234. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Clark PE, Polosukhina DA, Gyabaah K, Moses
HL, Thorburn A and Zent R: TRAIL and interferon-alpha act
synergistically to induce renal cell carcinoma apoptosis. J Urol.
184:1166–1174. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang M, He Z, Wen L, et al: Cadmium
suppresses the proliferation of piglet Sertoli cells and causes
their DNA damage, cell apoptosis and aberrant ultrastructure.
Reprod Biol Endocrinol. 8:972010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
He J and Bazan HE: Epidermal growth factor
synergism with TGF-beta1 via PI-3 kinase activity in corneal
keratocyte differentiation. Invest Ophthalmol Vis Sci.
49:2936–2945. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Merlo S, Frasca G, Canonico PL and Sortino
MA: Differential involvement of estrogen receptor alpha and
estrogen receptor beta in the healing promoting effect of estrogen
in human keratinocytes. J Endocrinol. 200:189–197. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Xu C, Gui Q, Chen W, et al: Small
interference RNA targeting tissue factor inhibits human lung
adenocarcinoma growth in vitro and in vivo. J Exp Clin Cancer Res.
30:632011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Cool CD, Stewart JS, Werahera P, et al:
Three-dimensional reconstruction of pulmonary arteries in plexiform
pulmonary hypertension using cell-specific markers. Evidence for a
dynamic and heterogeneous process of pulmonary endothelial cell
growth. Am J Pathol. 155:411–419. 1999. View Article : Google Scholar
|
|
32
|
Yi ES, Kim H, Ahn H, et al: Distribution
of obstructive intimal lesions and their cellular phenotypes in
chronic pulmonary hypertension. A morphometric and
immunohistochemical study. Am J Respir Crit Care Med.
162:1577–1586. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wang Q, Zuo XR, Wang YY, Xie WP, Wang H
and Zhang M: Monocrotaline-induced pulmonary arterial hypertension
is attenuated by TNF-α antagonists via the suppression of TNF-α
expression and NF-κB pathway in rats. Vascul Pharmacol. 58:71–77.
2013.
|
|
34
|
Fujita M, Shannon JM, Irvin CG, et al:
Overexpression of tumor necrosis factor-alpha produces an increase
in lung volumes and pulmonary hypertension. Am J Physiol Lung Cell
Mol Physiol. 280:L39–L49. 2001.PubMed/NCBI
|
|
35
|
Zhao YD, Courtman DW, Deng Y, Kugathasan
L, Zhang Q and Stewart DJ: Rescue of monocrotaline-induced
pulmonary arterial hypertension using bone marrow-derived
endothelial-like progenitor cells: efficacy of combined cell and
eNOS gene therapy in established disease. Circ Res. 96:442–450.
2005. View Article : Google Scholar
|
|
36
|
Sage E, Mercier O, Van den Eyden F, et al:
Endothelial cell apoptosis in chronically obstructed and reperfused
pulmonary artery. Respir Res. 9:192008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Taraseviciene-Stewart L, Kasahara Y, Alger
L, et al: Inhibition of the VEGF receptor 2 combined with chronic
hypoxia causes cell death-dependent pulmonary endothelial cell
proliferation and severe pulmonary hypertension. FASEB J.
15:427–438. 2001. View Article : Google Scholar
|
|
38
|
Teichert-Kuliszewska K, Kutryk MJ,
Kuliszewski MA, et al: Bone morphogenetic protein receptor-2
signaling promotes pulmonary arterial endothelial cell survival:
implications for loss-of-function mutations in the pathogenesis of
pulmonary hypertension. Circ Res. 98:209–217. 2006. View Article : Google Scholar
|
|
39
|
Sun CK, Lee FY, Sheu JJ, et al: Early
combined treatment with cilostazol and bone marrow-derived
endothelial progenitor cells markedly attenuates pulmonary arterial
hypertension in rats. J Pharmacol Exp Ther. 330:718–726. 2009.
View Article : Google Scholar
|
|
40
|
Liu Y, Ma C, Zhang Q, et al: The key role
of transforming growth factor-beta receptor I and 15-lipoxygenase
in hypoxia-induced proliferation of pulmonary artery smooth muscle
cells. Int J Biochem Cell Biol. 44:1184–1202. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ismail S, Sturrock A, Wu P, et al: NOX4
mediates hypoxia-induced proliferation of human pulmonary artery
smooth muscle cells: the role of autocrine production of
transforming growth factor-{beta}1 and insulin-like growth factor
binding protein-3. Am J Physiol Lung Cell Mol Physiol.
296:L489–L499. 2009.PubMed/NCBI
|
|
42
|
Revermann M, Barbosa-Sicard E, Dony E, et
al: Inhibition of the soluble epoxide hydrolase attenuates
monocrotaline-induced pulmonary hypertension in rats. J Hypertens.
27:322–331. 2009. View Article : Google Scholar : PubMed/NCBI
|