Introduction
Parkinson’s disease (PD) is characterized by the
progressive loss of dopaminergic neurons in the substantia nigra
pars compacta (SNpc) and their axon terminals in the striatum
(ST) associated with a complex, but slow onset of motor symptoms,
including bradykinesia, muscular rigidity, resting tremor and gait
abnormalities with poor postural balance (1). Monoamine oxidase type-B (MAO-B)
inhibitors have returned to the spotlight as an alternative to
dopaminergic replacement therapy and studies have demonstrated that
they enhance cognitive function (2) and exert neuroprotective effects
(3). Disease modification has
also been investigated in previous studies, assessing the effects
of the MAO-B inhibitors, selegiline and rasagiline (3,4).
Although selegiline, the first selective inhibitor of MAO-B, has
been widely used in patients with PD as monotherapy and adjuvant
therapy, its basic and clinical pharmacological effects have not
yet been fully elucidated. There is evidence that its
neuroprotective characteristics are mediated through its effects on
protein kinase C and mitogen-activated protein kinase signaling
pathways (5). Indeed, the
improvements observed as regards clinical PD progression following
the use of this type of drug have confirmed its neuroprotective
activities, which have been previously reported in various cell
culture and preclinical in vivo models (6–10).
The neuroprotective effects of MAO inhibitors also
involve the regulation of cell survival/death pathways, including
those involving Bcl-2 family proteins and the
glyceraldehyde-3-phosphate dehydrogenase (GAPDH) death cascade
(11,12). Unlike neuroprotective therapeutic
strategies, neurorescue or neurorestorative therapies aim to
eliminate neuronal deficits and degeneration after impairment
onset. Previous studies have reported that MAO-B inhibitors can
facilitate the availability of neurotrophic factors (NTFs) in
vitro, particularly glial cell line-derived neurotrophic factor
(GDNF) and brain-derived neurotrophic factor (BDNF) (13,14) and have demonstrated that these
outcomes have neurorestorative effects (15,16). However, to our knowledge, there
have been no investigations assessing the possible neurorestorative
effects of selegiline on behavioral deficits and molecular
alterations associated with NTFs in vivo. This gap in the
current understanding prompted us to perform experiments assessing
the possible neurorescue activity of selegiline and the underlying
mechanisms in a subacute
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced mouse
model of PD.
Materials and methods
Animal protocols
All procedures were approved by the Animal Ethics
Committee of Zhongshan Hospital, Fudan University, Shanghai, China
and carried out in accordance with the National Institutes of
Health Guide for the Care and Use of Laboratory Animals.
Experiments were conducted using 10-week-old male C57BL/6 mice
weighing 24–26 g purchased from Shanghai SLAC Laboratory Animal
Co., Ltd. (Shanghai, China). The animals were maintained in
standard conditions (12/12-h light/dark cycle, 21±2°C and relative
humidity of 40%) and allowed access to food and water ad
libitum.
Administration of MPTP and
selegiline
An MPTP model of PD was generated as previously
described (17,18). Briefly, the mice received daily
intraperitoneal (i.p.) injections of the vehicle (saline) or MPTP
(30 mg/kg/day; Sigma, St. Louis, MO, USA) dissolved in
physiological saline for 5 consecutive days to induce Parkinsonism.
Each treatment group included 10 mice. Selegiline
([(R)-(−)-N,2-dimethyl-N-2-propynylphenethylamine]; L-deprenyl; 1.0
mg/kg/day; Sigma) was dissolved in physiological saline and the pH
was adjusted to 7.4 before it was given via intragastric (i.g.)
administration. Selegiline or vehicle (saline) treatment commenced
72 h after the final MPTP administration and was administered daily
for 14 days. The experimental groups were as follows: group I,
normal saline (NS) (i.p.) + NS (i.g.); group II, MPTP (i.p.) + NS
(i.g.); group III, MPTP (i.p.) + selegiline 1.0 mg/kg/day (i.g.).
The mice were sacrificed by cervical dislocation or perfusion 24 h
after the final vehicle or selegiline administration.
Gait test
The gait test was performed according to previously
published methods (19–21) with minor modifications. The
apparatus was composed of a runway [dimensions: 4.5 cm (w) × 40 cm
(l) × 12 cm (h)] illuminated by a light (60 W), and a black wooden
box [20 cm (w) × 17 cm (l) × 10 cm (h)] was placed at one end of
the runway. The fore and hind paws of the animals were wet with
blue ink and they were allowed to trot on a strip of paper (4.5 cm
wide, 40 cm long) down the brightly lit runway towards the black
goal box. Stride lengths were manually measured as the distance
between 2 paw prints. The 3 longest stride lengths (corresponding
to maximal velocity) were measured from each run. Paw prints made
at the beginning (7 cm) and end (7 cm) of the run were excluded due
to changes in velocity. Runs in which the mice were observed making
stops or significant decelerations were excluded from the analysis.
The behavioral assessment was performed 3 days before the first
MPTP injection and on the 7th and 14th day of selegiline or vehicle
treatment.
Perfusion and tissue processing
At the end of the experiment, half of the animals
(n=5) in each group were sacrificed under 10% chloral hydrate
overdose anesthesia (360 mg/kg) then perfused via intracardial
infusion with saline (0.9%) followed by 4% paraformaldehyde (PFA),
pH 7.4. Following intracardial perfusion, the brains were collected
and post-fixed in 4% PFA for 24 h at 4°C, embedded in paraffin and
cut into 5-μm-thick coronal sections encompassing the entire SNpc
and ST (antero-posterior levels: −3.64 to −2.92 mm and +0.86 to
+0.02 mm) as previously described (22).
Another 5 animals in each group were sacrificed by
cervical dislocation and the tissue of their ventral midbrain was
dissected rapidly on ice, frozen in liquid nitrogen and stored at
−80°C until use.
Tyrosine hydroxylase (TH)
immunohistochemistry
Immunohistochemistry was performed as previously
described (23,24) with minor modifications. Briefly,
the fixed brain sections were incubated with 0.3% hydrogen peroxide
(H2O2) for 10 min at room temperature to
quench endogenous peroxidase activity and then placed in blocking
buffer containing 10% goat serum with 0.2% Triton X-100 in 0.01 M
phosphate-buffered saline (PBS; pH 7.2) for 30 min at 37°C. In each
treatment, the slides were washed at least 3 times with 0.01 M PBS
for 5 min each, followed by incubation at 4°C overnight with mouse
anti-TH monoclonal antibody (Sigma) at 1:2,000 dilution in 0.01 M
PBS containing 1% goat serum and 0.2% Triton X-100. The following
day, the sections were treated for 30 min with biotinylated
anti-mouse IgG and then processed with streptavidin-peroxidase
complex (ABC kit; Vector Laboratories, Burlingame, CA, USA). The
peroxidase reaction was visualized by 0.05% diaminobenzidine (DAB)
with 0.03% H2O2 in Tris-HCl buffer. Adjacent
sections were stained with cresyl violet to confirm cell
vitality.
Quantification of TH-immunoreactive
neurons and fibers
The number of dopaminergic neurons was determined as
previously described (25).
Briefly, we manually counted TH-positive cells under bright-field
illumination in the right SNpc using a ×10 or ×20 objective with a
DP71 camera (Olympus, Center Valley, PA, USA). Cell counts were
determined blindly on 5 anatomically matched sections from each of
the animals (n=5/group). It should be noted that the analyses of
the TH-immunoreactive profiles were restricted to the SNpc and thus
excluded the ventral tegmental area. In addition, neurons were only
counted if they contained a nucleus that was surrounded by
cytoplasm.
The optical density (OD) of the striatal
dopaminergic fibers was analyzed using Image-Pro Plus Software
(Media Cybernetics, Inc., Rockville, MD, USA), according to a
previously described optical dissector method (26,27). The average labeling for each area
was calculated on 5 anatomically matched brain sections
(n=5/group). For further determination of the number of TH-positive
axons in the ST, we selected the section corresponding to bregma
+0.260 mm at high magnification in a 45-μm2 area
according to a previously described method (25).
Real-time polymerase chain reaction
(PCR)
Total RNA was isolated by homogenizing frozen
ventral midbrain (left side) tissue in 1 ml TRIzol reagent
(Invitrogen, Carlsbad, CA, USA) followed by isopropanol
precipitation (n=5). The resulting pellets were washed with 70%
ethanol and suspended in RNase-free water and the concentration of
RNA was determined using a GeneQuant RNA/DNA Calculator (Amersham
Biosciences, Piscataway, NJ, USA). RNA (1 μg) was taken as a
template and total cDNA synthesis was performed using a
PrimeScript™ RT Reagent kit (Takara, Shiga, Japan). SYBR-Green PCR
[using SYBR Premix Ex Taq™ (Takara)] amplification was performed in
a Realplex4 S Real-time PCR instrument (Eppendorf, Hamburg,
Germany). β-actin was labeled with a reporter dye and used as an
endogenous control. The relative fold changes were determined using
the 2−ΔΔCt method as previously described (28). All primers (Table I) were designed according to the
relevant literature and synthesized by Genemed Biotechnologies,
Inc. (South San Francisco, CA, USA).
 | Table IPrimer sequences used for real-time
PCR. |
Table I
Primer sequences used for real-time
PCR.
| Gene name | Gene ID | | Primer
sequence |
|---|
| GDNF | NM_010275.2 | Sense: | 5′-AAG GTC ACC AGA
TAA ACA AGC GG-3′ |
| | Antisense: | 5′-TCA CAG GAG CCG
CTG CAA TAT C-3′ |
| BDNF | NM_007540.4 | Sense: | 5′-ACT ATG GTT ATT
TCA TAC TTC GGT T-3′ |
| | Antisense: | 5′-CCA TTC ACG CTC
TCC AGA-3′ |
| Bax | NM_007527.3 | Sense: | 5′-CGG CGA ATT GGA
GAT GAA CTG-3′ |
| | Antisense: | 5′-GCA AAG TAG AAG
AGG GCA ACC-3′ |
| Bcl-2 | NM_177410.2 | Sense: | 5′-ACC GTC GTG ACT
TCG CAG AG-3′ |
| | Antisense: | 5′-GGT GTG CAG ATG
CCG GTT CA-3′ |
| β-actin | NM_007393.3 | Sense: | 5′-CCT CTA TGC CAA
CAC AGT GC-3′ |
| | Antisense: | 5′-GTA CTC CTG CTT
GCT GAT CC-3′ |
Western blot analysis
For western blot analysis, isolated tissues from the
ventral midbrain (right side) were homogenized in RIPA buffer [50
mM Tris (pH 7.4), 150 mM NaCl, 1% Triton X-100, 1 % sodium
deoxycholate, 0.1% sodium dodecyl sulfate (SDS) and proteinase
inhibitors; Beyotime, Shanghai, China] and centrifuged at 13,000
rpm at 4°C for 5 min. Total protein content in the supernatant was
determined using a BCA Protein Assay kit (Beyotime) with a
spectrophotometer (Labomed, Inc., Culver City, CA, USA), and it was
diluted to an appropriate final concentration with homogenization
buffer and a protein solubilization solution. The sample was boiled
for 3 min and 30 μg of protein from each sample was electrophoresed
on a 10% SDS polyacrylamide gel then electrophoretically
transferred onto a nitrocellulose membrane in transfer buffer using
a Trans Blot SD apparatus (Bio-Rad, Hercules, CA, USA). The
membrane was then blocked by immersion in Tris-buffered saline
containing Tween-20 (TBST) and 1% BSA for 4 h at room temperature
and incubated at 4°C overnight with mouse primary antibodies:
anti-GDNF and anti-BDNF (1:200; Santa Cruz Biotechnology, Inc.,
Santa Cruz, CA USA) and anti-Bax, anti-Bcl-2 and anti-β-actin
(1:1,000; Santa Cruz Biotechnology, Inc.). After rinsing 3 times in
TBST for 10 min, blots were incubated for 2 h at room temperature
with an anti-rabbit IgG-peroxidase conjugated secondary antibody
(1:2,000; Santa Cruz Biotechnology, Inc.). Immunoreactivity was
visualized with an enhanced chemiluminescence detection system (GE
Healthcare, Piscataway, NJ, USA). The blots were scanned with a
KODAK In-Vivo Multispectral Imaging System FX (Carestream
Health, Rochester, NY, USA) during a 5-min exposure time and images
were automatically acquired with a CCD camera. The intensity of the
protein bands was measured by densitometry and expressed as a ratio
to β-actin intensity as previously described (29).
Terminal deoxynucleotidyl
transferase-mediated dUTP nick-end labeling (TUNEL) assay
TUNEL assays were performed according to previously
described methods (23,30) with minor modifications. Briefly,
an In Situ Death Detection kit (Roche, Basel, Switzerland)
was used according to the manufacturer’s instructions on serial
coronal brain sections encompassing the SNpc (bregma, −2.92−3.64
mm). The total number of TUNEL-positive cells within the SNpc was
counted in 14–15 slides/animal under a light microscope equipped
with a ×20 objective lens.
Statistical analyses
All data are presented as the means ± standard error
of mean (SEM). One-way analysis of variance (ANOVA) followed by
post-hoc analyses of Tukey’s honestly significant difference (HSD)
and Student-Newman-Keuls multiple comparisons tests were performed
using SPSS 16.0 software (SPSS, Inc., Chicago, IL, USA). A P-value
<0.05 was considered to indicate a statistically significant
difference. Linear regression analysis was applied to assess the
correlations between 2 parameters.
Results
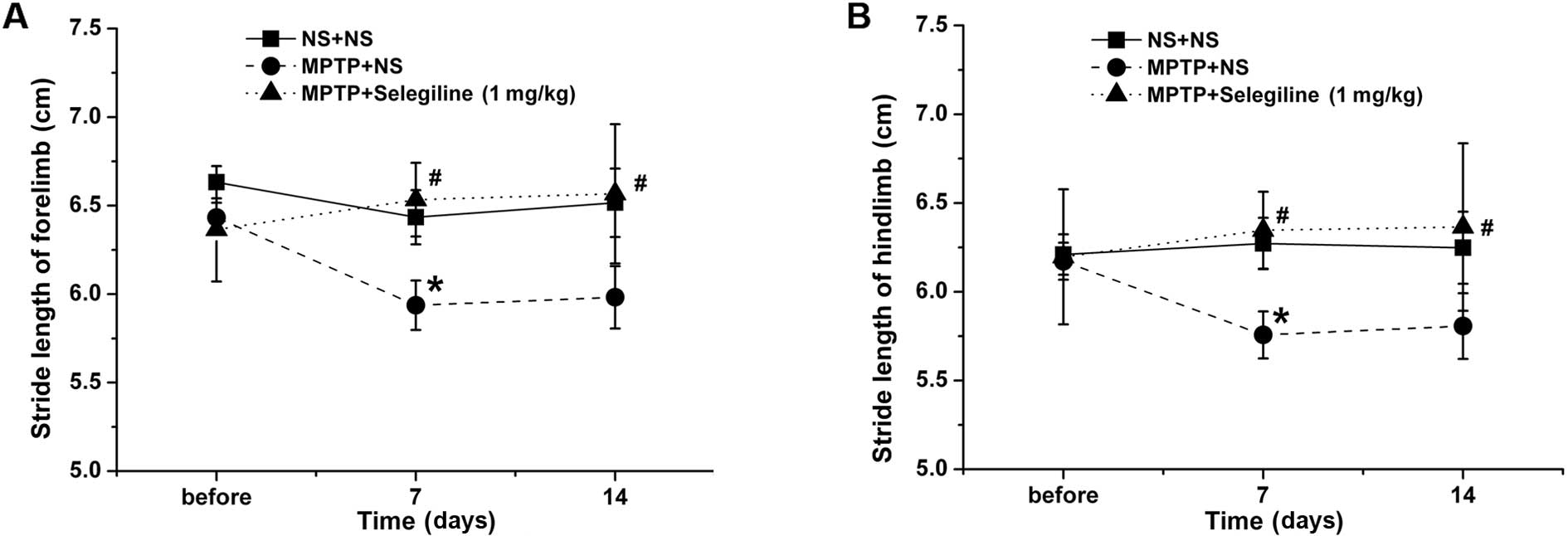
Selegiline improves gait dysfunction in a
subacute MPTP mouse model of PD
Shortened stride length is one of the chief
characteristics of abnormal gait in patients with PD (31). Accordingly, we observed a
significant decrease in fore- and hindlimb stride length in the
MPTP-exposed mice treated with vehicle (saline) on the 9th day
after the final MPTP administration (7th day of vehicle treatment),
compared with the normal control group (P=0.023 and P=0.014,
respectively) (Fig. 1). The fore-
and hindstride lengths of the mice in the selegiline (1.0
mg/kg/day) treatment group were longer than those in the
MPTP-vehicle group (P=0.024 and P=0.029, respectively). Improvement
in the selegiline-treated group was also observed on the 14th day
of treatment compared with the MPTP-exposed mice (P=0.032 and
P=0.044, respectively).
Selegiline attenuates the loss of
TH-positive nigral neurons and striatal axons in subacute
MPTP-exposed mice
After 14 days of treatment with selegiline or the
vehicle and the completion of the behavioral assessment, half of
the mice in each group were sacrificed and the brains were prepared
for TH-immunoreactivity experiments. Representative coronal
mesencephalon sections containing TH-positive neurons and fibers in
the SNpc and ST are shown in Fig.
2A–D. There was a significant decrease in the number of
TH-positive nigral dopaminergic neurons in the vehicle-treated,
MPTP-exposed group compared with the non-exposed control mice
(42.93% of saline control, P=0.000) (Fig. 2A, B and E). In the mice receiving
daily oral selegiline treatment, the number of TH-positive neurons
was significantly higher than that in the MPTP/vehicle-treated
animals (192.68% of MPTP control, P=0.001) and did not differ
compared with the non-exposed control mice (82.72% of saline
control, P>0.05).
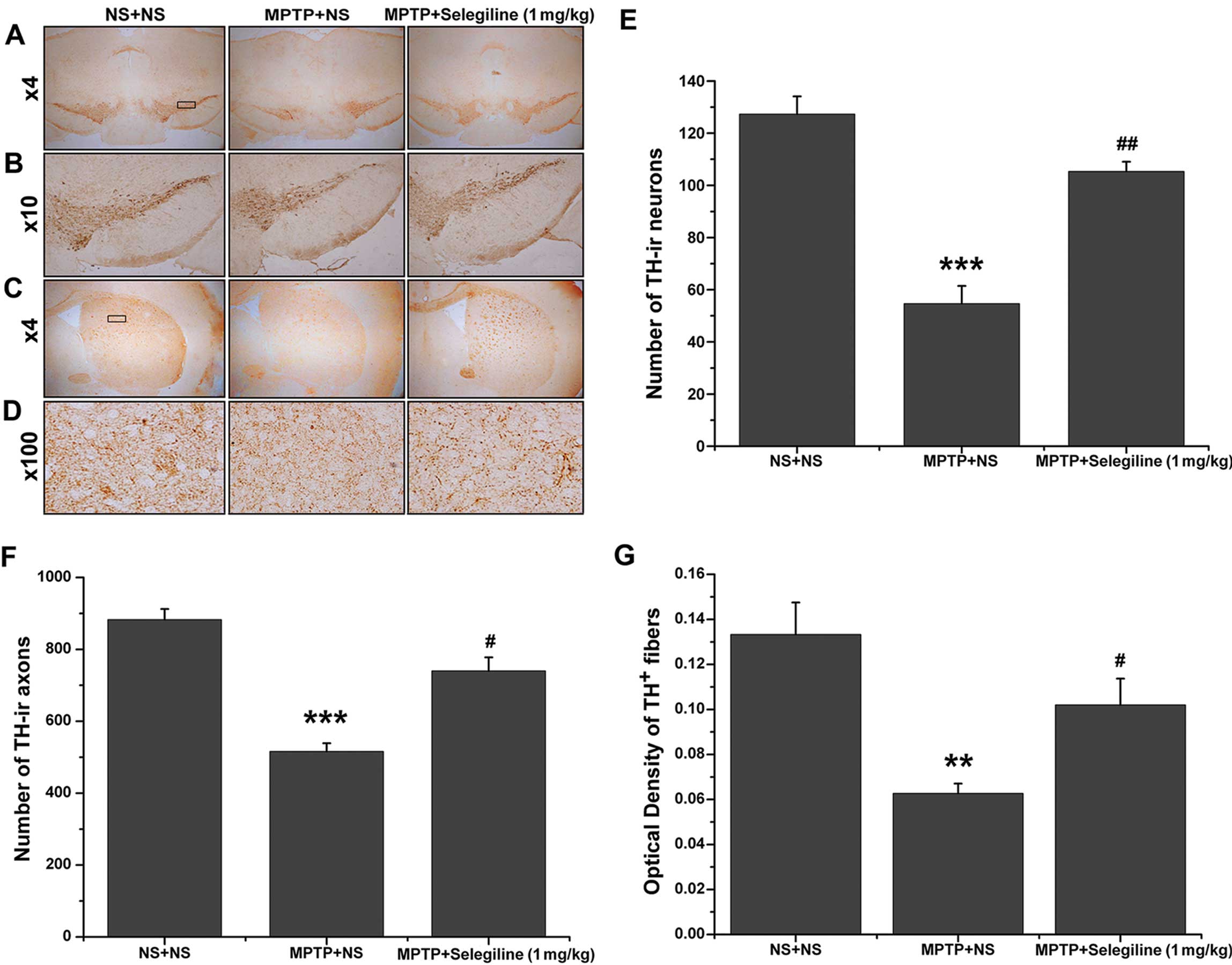 | Figure 2Neurorescue effects of selegiline
against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)
detected by tyrosine hydroxylase (TH) immunohistochemistry. (A and
B) Representative photomicrographs of TH-positive neurons in the
substantia nigra pars compacta (SNpc). (C and D)
Representative photomicrographs of TH-positive striatal fibers. (E)
Following MPTP injection, the number of TH-positive neurons in the
SNpc was significantly reduced; however, a marked recovery was
observed in the selegiline-treated (1.0 mg/kg/day) group compared
with the vehicle-treated MPTP-exposed mice. (F and G) MPTP
decreased the number of TH-positive axons and the optical density
(OD) of striatal fibers, which were preserved by selegiline
treatment. Data are expressed as the means ± SEM (n=5 each).
**P<0.01, ***P<0.001 vs. NS + NS group;
#P<0.05, ##P<0.01 vs. MPTP + NS group.
NS, normal saline; TH-ir axons, axons showing TH-like
immunoreactivity. |
We also observed a reduction in the number of
TH-positive axons and fibers throughout the dorsal ST of the
MPTP-exposed animals; however, this damage improved in the
selegiline-treated group. Both the number and OD analysis of
TH-positive fibers revealed a significant loss of dopamine (DA)
terminals in the MPTP/vehicle-treated group (58.44 and 47.37% of
saline control; P=0.000 and 0.001, respectively) (Fig. 2F and G). By contrast, the number
and density of TH-positive axons and fibers were clearly increased
in the MPTP-treated mice that received selegiline compared with
those that received saline (143.41 and 162.76% of MPTP control;
P=0.015 and 0.038, respectively) (Fig. 2C, D, F and G), bringing them to
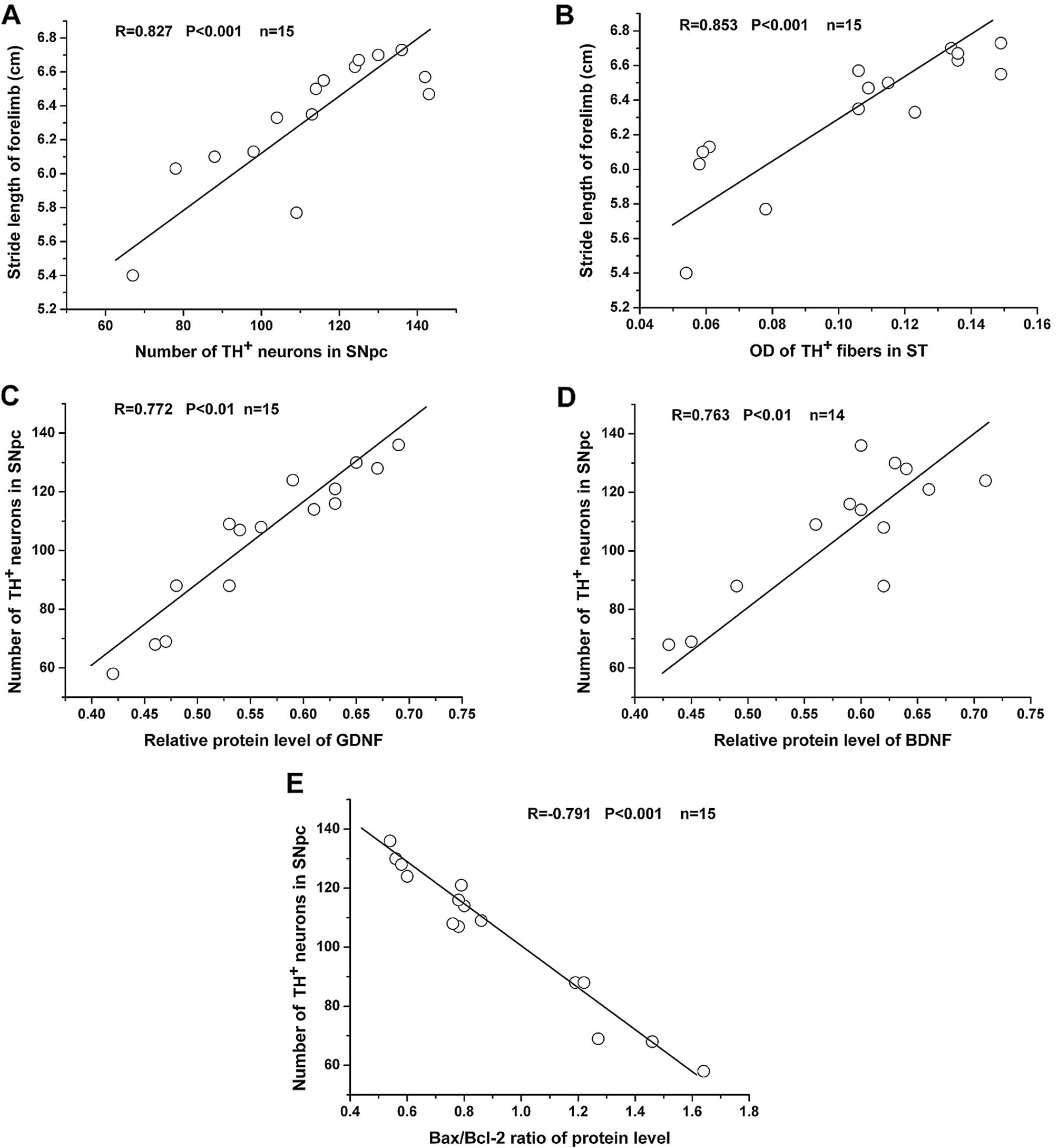
83.90 and 76.69% of the normal control levels (P>0.05). Linear
regression analysis revealed that there was a strong positive
correlation between forelimb stride length and the number of
TH-positive SNpc neurons, as well as the OD of TH-positive striatal
fibers (Fig. 5A and B).
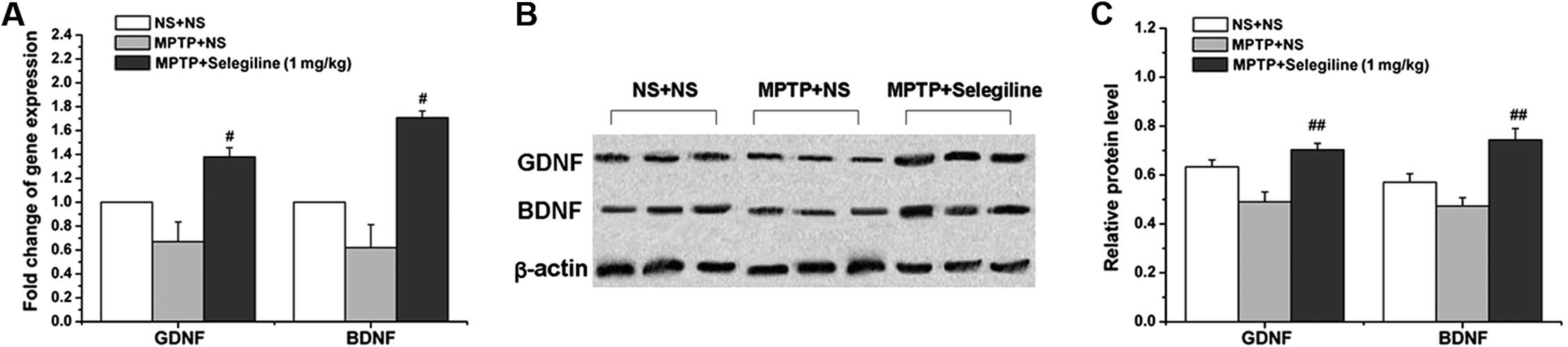
Selegiline increases the relative mRNA
and protein levels of GDNF and BDNF in the SNpc of subacutely
MPTP-exposed mice
We performed real-time PCR and western blot analyses
to assess changes in GDNF and BDNF expressions at the mRNA and
protein level following treatment with selegiline. We observed a
significant increase in the relative mRNA and protein levels of
GDNF in the MPTP/selegiline-treated animals compared with the
MPTP/vehicle-treated mice (2.10-fold in mRNA and 143.53% in protein
of MPTP control; P=0.017 and 0.009, respectively). There were
similar changes in BDNF expression; we observed significantly
higher relative mRNA and protein levels in the
MPTP/selegiline-treated group compared with the
MPTP/vehicle-treated animals (2.75-fold and 157.05% of MPTP
control; P=0.048 and 0.004, respectively) (Fig. 3A–C). These results demonstrate
that selegiline induces the gene and protein expression of GDNF and
BDNF. Linear regression analysis revealed that there was a strong
positive correlation between GDNF/BDNF protein levels and the
number of TH-positive SNpc neurons (Fig. 5 C and D).
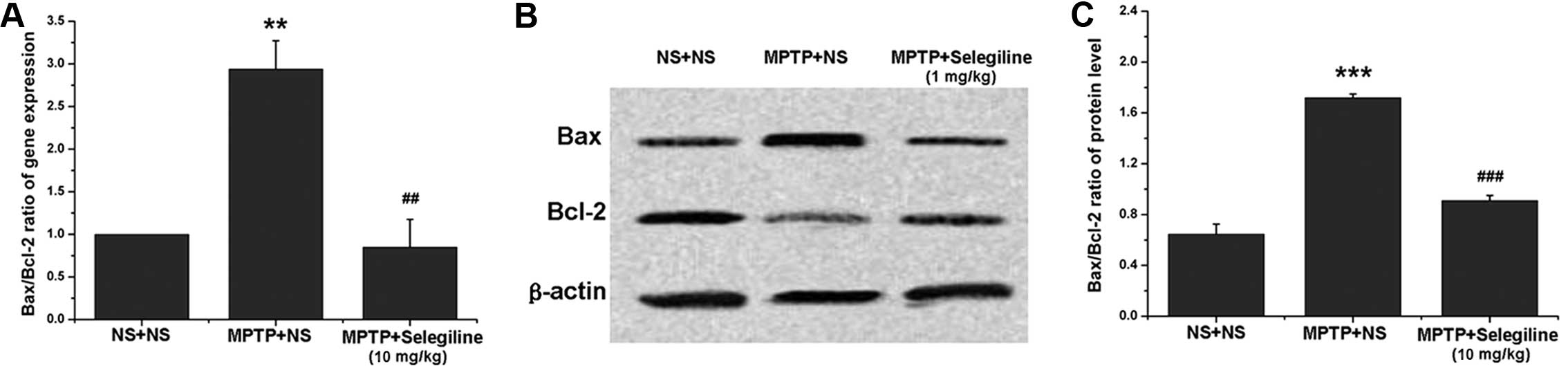
Selegiline attenuates the relative mRNA
and protein ratios of Bax/Bcl-2 in the SNpc of subacutely
MPTP-exposed mice
The effects of selegiline on apoptosis were assessed
by analyzing Bax and Bcl-2 expression by real-time PCR and western
blot analyses. The relative mRNA level of the pro-apoptotic factor,
Bax, increased in the ventral midbrain of MPTP-exposed mice
(2.14-fold of saline control, P=0.037), while that of the
anti-apoptotic factor, Bcl-2, did not differ significantly between
the treated mice and the normal control mice (P>0.05). However,
the mRNA ratio of Bax/Bcl-2 increased significantly in the
MPTP-exposed mice (1.99-fold of saline control, P=0.002) (Fig. 4A); however, this increase was
reversed within 14 days of selegiline treatment (58.79% of MPTP
control, P=0.004) (Fig. 4A).
Similarly, the protein ratio of Bax/Bcl-2 significantly increased
in the MPTP-exposed mice (265.97% of saline control, P=0.000), and
this increase was reversed by selegiline (52.91% of MPTP control,
P=0.000) (Fig. 4B and C), due to
an obvious downregulation of Bax (73.70% of MPTP control, P=0.023)
and an upregulation of Bcl-2 (140.38% of MPTP control, P=0.001).
Linear regression analysis revealed a strong negative correlation
between the Bax/Bcl-2 protein ratio and the number of TH-positive
SNpc neurons (Fig. 5E).
Selegiline effectively reverses apoptosis
in the SNpc of MPTP-treated animals
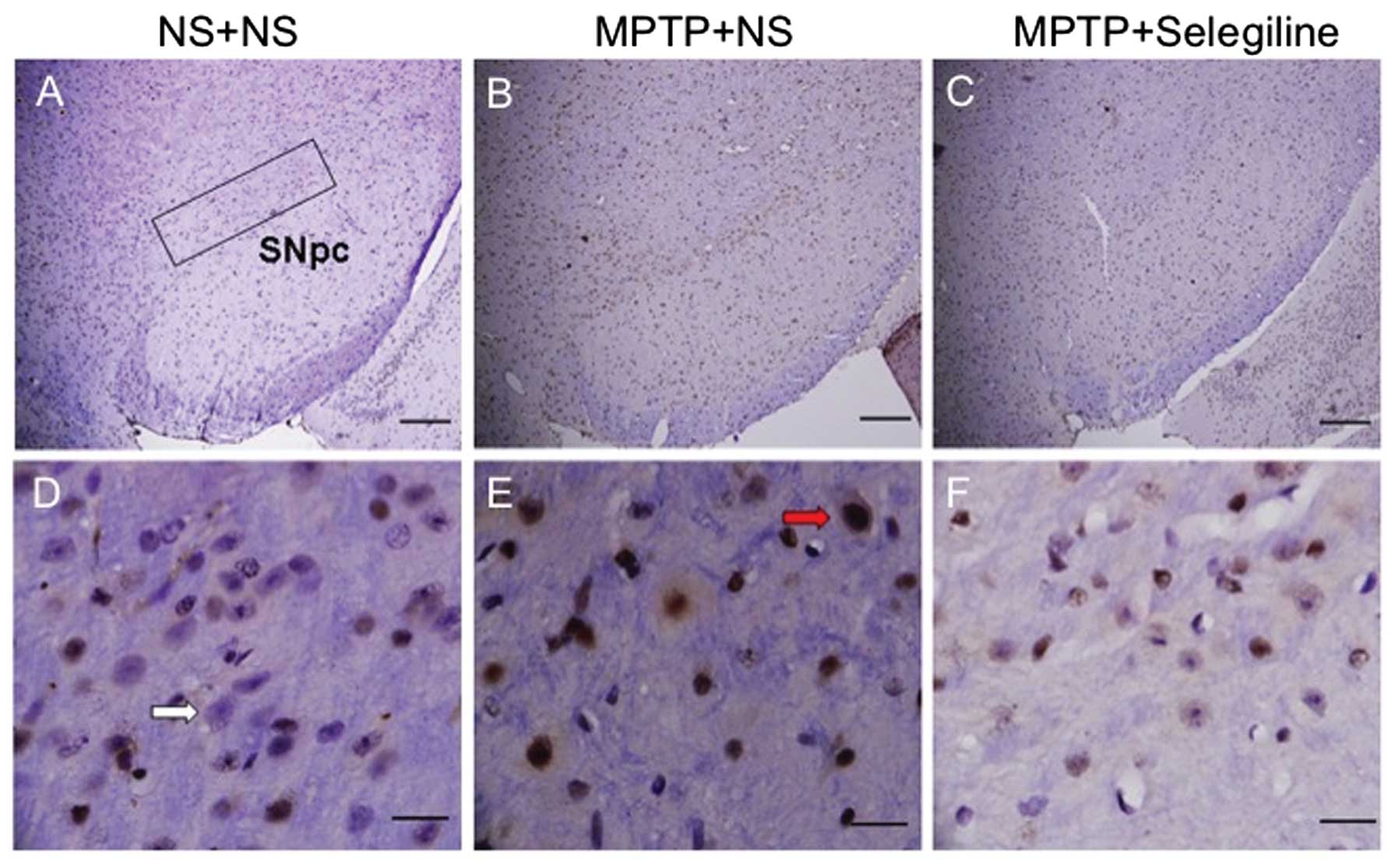
We performed TUNEL staining (Fig. 6) and observed that the SNpc of the
MPTP/vehicle-treated animals contained more apoptotic nuclei
(Fig. 6E, red arrow) than the
control animals. Notably, the MPTP/selegiline-treated mice did not
show any evidence of apoptosis (Fig.
6F).
Discussion
Our results demonstrate that selegiline, the first
MAO-B inhibitor, rescues motor deficits and induces NTF expression
in a subacute MPTP mouse model of PD, which is the most commonly
used model of PD. The magnitude of the MPTP-induced lesion is
dependent on the administration regimen (17). The subacute regimen induces a
40–50% depletion of striatal DA levels and a 30–40% SNpc neuronal
loss (18). Compared to the more
severe acute regimen, the subacute regimen was more appropriate for
our experiments, in which we sought to identify neurorestorative
effects.
We observed that 14 days of oral selegiline restored
the number of nigral dopaminergic neurons, the number and density
of striatal dopaminergic terminals and improved gait dysfunction
compared with the vehicle/MPTP-treated mice. Moreover, our results
suggest that the neurorescue effects of selegiline are mediated by
the induction of GDNF and BDNF expression, as well its regulatory
effects on Bax and Bcl-2, 2 key molecules of the Bcl-2 family
involved in the apoptosis of dopaminergic neurons in PD
pathogenesis.
MAO-B inhibition is known to diminish the rapid
turnover of striatal DA, allowing for it to accumulate. For a
patient with PD, blocking endogenous DA catabolism provides
symptomatic relief through enhanced neurotransmission (32). Increasing endogenous DA
concentrations may be a practical alternative to dopaminergic
replacement therapy (33).
Clinical studies have shown that compared with dopaminergic
therapy, MAO-B inhibitors, including selegiline and rasagiline,
offer limited symptomatic improvement when administered as
monotherapy (34–36). Thus, it remains unclear whether
the motor effects of selegiline are associated with MAO-B
inhibition or with its neuroprotective activities. We noted a
decrease in stride length in the MPTP-exposed mice, similar to the
characteristic shuffling gait in patients with PD. Indeed, this
results from a combination of hypokinesia, rigidity and posture and
equilibrium defects. However, post-treatment selegiline reversed
the shortening of the stride lengths. Moreover, this was not
associated with other neurorescue mechanisms apart from MAO-B
inhibition; the dose of selegiline (1.0 mg/kg/day) used in our
study was lower than the dose reported to inhibit MAO (37). Furthermore, the delayed start of
administration in our experiment ensured that any observed effects
were not due to the compound interfering with the conversion of
MPTP to its active metabolite, MPP+, a reaction that is
mediated by MAO-B (15). Previous
studies have demonstrated that selegiline protects against
MPP+ toxicity, even in cell lines that lack MAO-B
(38,39). Moreover, MAO-B-knockout mice are
not protected from damage caused by hypoxia or MPP+
(40). In short, the neurorescue
effects on gait dysfunction observed suggest novel molecular
mechanisms of action of selegiline that are independent of MAO-B
inhibition.
Another significant effect of selegiline was the
recovery of TH-immunopositive neurons and fibers in the
MPTP-exposed mice. This finding is similar with the results of
previous studies on rasagiline, a second-generation irreversible,
selective MAO-B inhibitor. However, the effects of rasagiline on
striatal DA content did not correlate with its MAO-B inhibitory
activity (41). Proteomic and
genomic methods subsequently demonstrated that rasagiline induced
the activation of cell signaling mediators associated with an
NTF-responsive tyrosine kinase receptor (Trk) pathway and a
downstream increase of phosphatidylinositol 3 kinase (PI3K)
protein. The induction of NTFs, such as GDNF and BDNF seems to be
associated with the neurorescue mechanism(s) of rasagiline
(41). As regards selegiline,
‘trophic-like’ action or NTF induction has been reported in both
in vitro and in vivo neuroprotective studies
(13,42). Our data demonstrate the rescue
effects of low-dose selegiline on dopaminergic neurons and fiber
loss in MPTP-exposed mice and confirm that this subacute MAO
inhibitory dose also induces GDNF and BDNF mRNA and protein
expression, even after neuronal cell death has begun. These results
support and extend those of previous studies, showing that both the
gene and protein expression of several Trk-ligands (including GDNF
and BDNF) are induced by selegiline and rasagiline. Moreover, they
demonstrate the involvement of GDNF and BDNF in neurorescue or
restorative treatment for neurodegenerative diseases, particularly
PD. In our study, both the GDNF and BDNF protein levels were
significantly positively correlated with the number of TH-positive
SNpc neurons, which suggests that NTF reduction may play a role in
pathological changes underlying PD and suggests that increasing NTF
levels may be a useful therapeutic strategy.
Selegiline also increased neuronal survival by
interfering with the apoptotic signaling pathway, independent of
MAO-B inhibition. Previous studies have indicated that the
neuroprotective effects of selegiline are associated with the
decreased synthesis of pro-apoptotic proteins, such as Bax, c-jun
and GAPDH, and the increased synthesis of anti-apoptotic proteins,
such as Bcl-2, Cu-Zn superoxide dismutase and heat shock protein 70
(42). Bcl-2 pro-apoptotic family
members are known to participate in neuronal death in a variety of
PD models (43) and the ratio of
Bax/Bcl-2 is used to determine whether cells will live or die
(44). Thus, we investigated
anti-apoptotic signaling in the subacute MPTP mouse model, in which
dopaminergic neurodegeneration occurs through apoptosis. Similar
with pre-treatment studies on selegiline (42), we found that the
post-administration of selegiline inhibited the increase in the
Bax/Bcl-2 ratio at the gene and protein level compared with the
untreated MPTP-exposed group. In addition, the strong negative
correlation between the Bax/Bcl-2 protein ratio with the number of
TH-positive neurons further confirmed the involvement of Bcl-2
family members in the pathogenesis of a subacute MPTP-induced mouse
model of PD. TUNEL assays further demonstrated that selegiline
successfully prevented apoptosis, even when administered after
MPTP. Although it remains unclear whether there are common pathways
with respect to the correlation between Bcl-2 family members and
NTF expression, our current results suggest that increasing
endogenous GDNF and/or BDNF levels and regulating the expression of
Bcl-2 pro-apoptotic family members may be a useful strategy for
neuronal rescue therapies.
Finally, it seems prudent to discuss the clinical
implications for the observed improvement in gait dysfunction in
our study and the possible correlation with NTF induction. The
effects of selegiline on gait dysfunction in the MPTP mouse model
are not dependent on its MAO inhibitory effect, which is currently
the focus of promising clinical investigations. In PD, motor
symptoms, such as bradykinesia and rigidity respond well to DA
replacement medications. Although balance and gait problems may
also be reversed by dopaminergic agents early in the course of the
disease, they usually become resistant to these therapies as the
disease progresses (45). The
effects of selegiline on gait dysfunction and the significantly
positive correlation between stride length and pathological
characteristics in our study further support the efficacy of the
compound on PD-related motor dysfunction. Indeed, several studies
have reported that NTFs delay neuronal degeneration and the
progression of abnormal gait or walking patterns in rats (46–48). In patients with PD, intraputaminal
GDNF infusion resulted in the significant, sustained improvement of
bilateral motor functions, including gait and balance (49). These findings are in agreement
with our presumption that selegiline ameliorates gait impairment
and rescues the loss of dopaminergic neurons, mostly likely through
the induction of GDNF and BDNF expression.
In conclusion, the present study demonstrates that
selegiline exerts neurorescue effects on MPTP-induced gait
dysfunction and the loss of dopaminergic neurons and fibers in
vivo. These effects appear to correlate with the multifactorial
activities of this compound, including the enhancement of GDNF and
BDNF expression levels and the suppression of apoptosis in the
ventral midbrain of a subacute MPTP-exposed mouse model through the
regulation of Bcl-2 family members. Combined with the results of
previous in vitro and in vivo studies regarding the
neuroprotective effects of selegiline, we further demonstrate the
efficacy of selegiline in delaying PD symptom progression and
reversing existing neurodegenerative damage, even at a dose that
does not inhibit MAO-B.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (No. 81202814), Training Scheme
of Back-up Experts of Shanghai University of Traditional Chinese
Medicine (No. B-X-53) and the Medical Leader sponsorship by
Shanghai Municipal Government (No. 2007-057). We thank Professor
Fang Huang and Professor Danian Zhu for their guidance regarding
the experiments and manuscript.
Abbreviations:
|
MPTP
|
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
|
|
MAO-B
|
monoamine oxidase type-B
|
|
NTFs
|
neurotrophic factors
|
|
GDNF
|
glial cell line-derived neurotrophic
factor
|
|
BDNF
|
brain-derived neurotrophic factor
|
|
GAPDH
|
glyceraldehyde-3-phosphate
dehydrogenase
|
|
TH
|
tyrosine hydroxylase
|
|
SNpc
|
substantia nigra pars
compacta
|
|
ST
|
striatum
|
|
DA
|
dopamine
|
References
|
1
|
Lees AJ, Hardy J and Revesz T: Parkinson’s
disease. Lancet. 373:2055–2066. 2009.
|
|
2
|
Stocchi F, Vacca L, Grassini P, et al:
Symptom relief in Parkinson disease by safinamide: Biochemical and
clinical evidence of efficacy beyond MAO-B inhibition. Neurology.
67:S24–S29. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bar-Am O, Weinreb O, Amit T and Youdim MB:
The neuroprotective mechanism of 1-(R)-aminoindan, the major
metabolite of the anti-parkinsonian drug rasagiline. J Neurochem.
112:1131–1137. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Olanow CW, Hauser RA, Jankovic J, et al: A
randomized, double-blind, placebo-controlled, delayed start study
to assess rasagiline as a disease modifying therapy in Parkinson’s
disease (the ADAGIO study): rationale, design, and baseline
characteristics. Mov Disord. 23:2194–2201. 2008.
|
|
5
|
Weinreb O, Mandel S, Bar-Am O, et al:
Multifunctional neuroprotective derivatives of rasagiline as
anti-Alzheimer’s disease drugs. Neurotherapeutics. 6:163–174.
2009.PubMed/NCBI
|
|
6
|
Chen JJ and Ly AV: Rasagiline: a
second-generation monoamine oxidase type-B inhibitor for the
treatment of Parkinson’s disease. Am J Health Syst Pharm.
63:915–928. 2006.
|
|
7
|
Magyar K, Pálfi M, Jenei V and Szöko E:
Deprenyl: from chemical synthesis to neuroprotection. J Neural
Transm Suppl. 143–156. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Weinreb O, Amit T, Bar-Am O, Sagi Y,
Mandel S and Youdim MB: Involvement of multiple survival signal
transduction pathways in the neuroprotective, neurorescue and APP
processing activity of rasagiline and its propargyl moiety. J
Neural Transm Suppl. 457–465. 2006. View Article : Google Scholar
|
|
9
|
Youdim MB, Maruyama W and Naoi M:
Neuropharmacological, neuroprotective and amyloid precursor
processing properties of selective MAO-B inhibitor antiparkinsonian
drug, rasagiline. Drugs Today (Barc). 41:369–391. 2005. View Article : Google Scholar
|
|
10
|
Youdim MB and Tipton KF: Rat striatal
monoamine oxidase-B inhibition by l-deprenyl and rasagiline: its
relationship to 2-phenylethylamine-induced stereotypy and
Parkinson’s disease. Parkinsonism Relat Disord. 8:247–253.
2002.PubMed/NCBI
|
|
11
|
Hara MR, Thomas B, Cascio MB, et al:
Neuroprotection by pharmacologic blockade of the GAPDH death
cascade. Proc Natl Acad Sci USA. 103:3887–3889. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Maruyama W, Akao Y, Carrillo MC, Kitani K,
Youdium MB and Naoi M: Neuroprotection by propargylamines in
Parkinson’s disease: suppression of apoptosis and induction of
prosurvival genes. Neurotoxicol Teratol. 24:675–682. 2002.
|
|
13
|
Maruyama W, Nitta A, Shamoto-Nagai M, et
al: N-Propargyl-1 (R)-aminoindan, rasagiline, increases glial cell
line-derived neurotrophic factor (GDNF) in neuroblastoma SH-SY5Y
cells through activation of NF-kappaB transcription factor.
Neurochem Int. 44:393–400. 2004. View Article : Google Scholar
|
|
14
|
Weinreb O, Bar-Am O, Amit T,
Chillag-Talmor O and Youdim MB: Neuroprotection via pro-survival
protein kinase C isoforms associated with Bcl-2 family members.
FASEB J. 18:1471–1473. 2004.PubMed/NCBI
|
|
15
|
Sagi Y, Mandel S, Amit T and Youdim MB:
Activation of tyrosine kinase receptor signaling pathway by
rasagiline facilitates neurorescue and restoration of nigrostriatal
dopamine neurons in post-MPTP-induced parkinsonism. Neurobiol Dis.
25:35–44. 2007. View Article : Google Scholar
|
|
16
|
Zhu W, Xie W, Pan T, et al: Comparison of
neuroprotective and neurorestorative capabilities of rasagiline and
selegiline against lactacystin-induced nigrostriatal dopaminergic
degeneration. J Neurochem. 105:1970–1978. 2008. View Article : Google Scholar
|
|
17
|
Jackson-Lewis V and Przedborski S:
Protocol for the MPTP mouse model of Parkinson’s disease. Nat
Protoc. 2:141–151. 2007.
|
|
18
|
Perier C, Bové J, Wu DC, et al: Two
molecular pathways initiate mitochondria-dependent dopaminergic
neurodegeneration in experimental Parkinson’s disease. Proc Natl
Acad Sci USA. 104:8161–8166. 2007.
|
|
19
|
Fernagut PO, Diguet E, Labattu B and Tison
F: A simple method to measure stride length as an index of
nigrostriatal dysfunction in mice. J Neurosci Methods. 113:123–130.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Fleming SM, Salcedo J, Fernagut PO, et al:
Early and progressive sensorimotor anomalies in mice overexpressing
wild-type human alpha-synuclein. J Neurosci. 24:9434–9440. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tillerson JL, Caudle WM, Reverón ME and
Miller GW: Detection of behavioral impairments correlated to
neurochemical deficits in mice treated with moderate doses of
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Exp Neurol.
178:80–90. 2002. View Article : Google Scholar
|
|
22
|
Paxinos G and Franklin KBJ: The Mouse
Brain in Stereotaxic Coordinates. 4th edition. Elsevier Academic
Press; San Diego: 2012
|
|
23
|
He XJ, Yamauchi H, Uetsuka K and Nakayama
H: Neurotoxicity of MPTP to migrating neuroblasts: studies in acute
and subacute mouse models of Parkinson’s disease. Neurotoxicology.
29:413–420. 2008.PubMed/NCBI
|
|
24
|
Yokoyama H, Takagi S, Watanabe Y, Kato H
and Araki T: Role of reactive nitrogen and reactive oxygen species
against MPTP neurotoxicity in mice. J Neural Transm. 115:831–842.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kuhn K, Wellen J, Link N, Maskri L,
Lübbert H and Stichel CC: The mouse MPTP model: gene expression
changes in dopaminergic neurons. Eur J Neurosci. 17:1–12. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
West MJ and Gundersen HJ: Unbiased
stereological estimation of the number of neurons in the human
hippocampus. J Comp Neurol. 296:1–22. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wu DC, Jackson-Lewis V, Vila M, et al:
Blockade of microglial activation is neuroprotective in the
1-methyl-4-phenyl-1,2, 3,6-tetrahydropyridine mouse model of
Parkinson disease. J Neurosci. 22:1763–1771. 2002.PubMed/NCBI
|
|
28
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. methods. 25:402–408. 2001.
|
|
29
|
Matsunaga W, Isobe K and Shirokawa T:
Involvement of neurotrophic factors in aging of noradrenergic
innervations in hippocampus and frontal cortex. Neurosci Res.
54:313–318. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Novikova L, Garris BL, Garris DR and Lau
YS: Early signs of neuronal apoptosis in the substantia nigra
pars compacta of the progressive neurodegenerative mouse
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine/probenecid model of
Parkinson’s disease. Neuroscience. 140:67–76. 2006.PubMed/NCBI
|
|
31
|
Blin O, Ferrandez AM and Serratrice G:
Quantitative analysis of gait in Parkinson patients: increased
variability of stride length. J Neurol Sci. 98:91–97. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Muller T: Drug therapy in patients with
Parkinson’s disease. Transl Neurodegener. 1:102012.
|
|
33
|
Lewitt PA: MAO-B inhibitor know-how: back
to the pharm. Neurology. 72:1352–1357. 2009. View Article : Google Scholar
|
|
34
|
Parkinson Study Group. DATATOP: a
multicenter controlled clinical trial in early Parkinson’s disease.
Arch Neurol. 46:1052–1060. 1989.
|
|
35
|
Parkinson Study Group. A controlled trial
of rasagiline in early Parkinson disease: the TEMPO study. Arch
Neurol. 59:1937–1943. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Olanow CW, Rascol O, Hauser R, Feigin PD,
Jankovic J, Lang A, Langston W, Melamed E, Poewe W, Stocchi F and
Tolosa E; ADAGIO Study Investigators. A double-blind, delayed-start
trial of rasagiline in Parkinson’s disease. N Engl J Med.
361:1268–1278. 2009.
|
|
37
|
Ansari KS, Yu PH, Kruck TP and Tatton WG:
Rescue of axotomized immature rat facial motoneurons by
R(−)-deprenyl: stereospecificity and independence from monoamine
oxidase inhibition. J Neurosci. 13:4042–4053. 1993.
|
|
38
|
Sharma SK, Carlson EC and Ebadi M:
Neuroprotective actions of Selegiline in inhibiting 1-methyl,
4-phenyl, pyridinium ion (MPP+)-induced apoptosis in
SK-N-SH neurons. J Neurocytol. 32:329–343. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Tatton WG, Chalmers-Redman RM, Ju WJ, et
al: Propargylamines induce antiapoptotic new protein synthesis in
serum- and nerve growth factor (NGF)-withdrawn, NGF-differentiated
PC-12 cells. J Pharmacol Exp Ther. 301:753–764. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Holschneider DP, Chen K, Seif I and Shih
JC: Biochemical, behavioral, physiologic, and neurodevelopmental
changes in mice deficient in monoamine oxidase A or B. Brain Res
Bull. 56:453–462. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Weinreb O, Amit T, Bar-Am O and Youdim MB:
Induction of neurotrophic factors GDNF and BDNF associated with the
mechanism of neurorescue action of rasagiline and ladostigil: new
insights and implications for therapy. Ann NY Acad Sci.
1122:155–168. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ebadi M, Sharma S, Shavali S and El Refaey
H: Neuroprotective actions of selegiline. J Neurosci Res.
67:285–289. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Levy OA, Malagelada C and Greene LA: Cell
death pathways in Parkinson’s disease: proximal triggers, distal
effectors, and final steps. Apoptosis. 14:478–500. 2009.
|
|
44
|
Tanaka K, Asanuma M and Ogawa N: Molecular
basis of anti-apoptotic effect of immunophilin ligands on hydrogen
peroxide-induced apoptosis in human glioma cells. Neurochem Res.
29:1529–1536. 2004. View Article : Google Scholar
|
|
45
|
Meredith GE and Kang UJ: Behavioral models
of Parkinson’s disease in rodents: a new look at an old problem.
Mov Disord. 21:1595–1606. 2006.
|
|
46
|
Patel M, Mao L, Wu B and Vandevord PJ:
GDNF-chitosan blended nerve guides: a functional study. J Tissue
Eng Regen Med. 1:360–367. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Piquilloud G, Christen T, Pfister LA,
Gander B and Papaloïzos MY: Variations in glial cell line-derived
neurotrophic factor release from biodegradable nerve conduits
modify the rate of functional motor recovery after rat primary
nerve repairs. Eur J Neurosci. 26:1109–1117. 2007. View Article : Google Scholar
|
|
48
|
Willson ML, McElnea C, Mariani J, Lohof AM
and Sherrard RM: BDNF increases homotypic olivocerebellar
reinnervation and associated fine motor and cognitive skill. Brain.
131:1099–1112. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Slevin JT, Gerhardt GA, Smith CD, Gash DM,
Kryscio R and Young B: Improvement of bilateral motor functions in
patients with Parkinson disease through the unilateral
intraputaminal infusion of glial cell line-derived neurotrophic
factor. J Neurosurg. 102:216–222. 2005. View Article : Google Scholar
|